How anticoags work, and what that means for labs

| How anticoags work, and what that means for labs |
|
|
|
January 2013 When to monitor and how? These questions were simpler when the list of anticoagulants patients were given was two or three deep—warfarin and unfractionated heparin, and maybe low-molecular-weight heparin. But the number of anticoagulants has grown and so has the complexity. Old and new—Michael Laposata, MD, PhD, covered the major anticoagulants and antiplatelet drugs in a recent AACC webinar and again at the CAP ’12 annual meeting in September. Here, this month, is an edited transcript of what he said in the webinar about warfarin and heparin and briefly about the newer drugs. In the February issue: a close-up look at fondaparinux, bivalirudin, rivaroxaban, dabigatran, lepirudin, and argatroban. Dr. Laposata is the Edward and Nancy Fody professor of pathology; executive vice chair of pathology, microbiology, and immunology and professor of medicine, Vanderbilt University School of Medicine; and pathologist-in-chief, Vanderbilt University Hospital, Nashville. Anticoagulants are dangerous drugs. If too much is given, the patient could bleed and bleed to death; if too little is given, the clot that was an inch long and sitting by the ankle suddenly starts growing up toward the knee. Once it gets above the knee, it has about a 50-50 chance to break off and enter the lung as a pulmonary embolism.
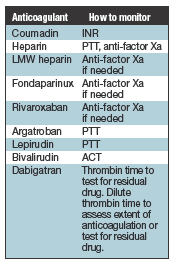
Anticoagulants are often prescribed along with a drug that inhibits the platelets. For example, in a patient with crushing chest pain, the aim is to prevent a clot over the plaque in the coronary artery that blocks the flow of blood and produces injury to the heart wall. There is concern about bleeding with both anticoagulants and antiplatelet drugs, but when used alone the antiplatelet agents are less likely to cause bleeding than the anticoagulants. The anticoagulant story used to be as short as warfarin and unfractionated heparin. But then heparin got a haircut, so to speak, and became low-molecular-weight heparin. Someone then figured out how to make small pieces of ‘heparin-like’ hair in a pharmaceutical laboratory and that is fondaparinux, which is a very short version that resembles heparin. Several other drugs came next: lepirudin, argatroban, bivalirudin, dabigatran, rivaroxaban. Dabigatran (Pradaxa) and rivaroxaban (Xarelto) are the newest ones added to the group. We have to understand all of these drugs, and we have to understand what to do in the clinical laboratory for a patient who is taking any one of them. Anticoagulant drugs are principally effective for venous thrombosis and preventing stroke in patients with atrial fibrillation. And there are other uses, such as for patients who have mechanical heart valves. Antiplatelet drugs are the mainstays for clots on the arterial side. Bleeding into the brain is a complication of major concern of anticoagulant therapy, and this is what we’re especially trying to avoid by balancing the amount of anticoagulant in the circulation. One of the problems driving the need for knowledge about the different anticoagulants is that the anesthesiologists and surgeons are told by patients needing surgery that they’re on compounds the anesthesiologists and surgeons may not have heard of. Consider the numerous following scenarios. Patients with atrial fibrillation could be on warfarin, dabigatran, or rivaroxaban. Patients with recurrent venous thrombosis typically are on warfarin but might be on heparin or low-molecular-weight heparin. Patients with heart valve replacements are on warfarin. The patient with acute coronary syndrome could be on heparin, warfarin, or low-molecular-weight heparin, in addition to a platelet inhibitor. Patients who have had a stroke might be treated with aspirin or anticoagulants. And many of these anticoagulants—warfarin, heparin and LMW heparin, fondaparinux, dabigatran, rivaroxaban—can be used prophylactically to prevent a clot. The surgery and anesthesiology groups at Vanderbilt are very interested in the laboratory’s clinical advice when deciding if a patient should have surgery or not. We are serving the OR with a coagulation laboratory on a cart. We do it now for liver transplant and plan to do it soon for cardiac surgery. On this cart are many coagulation assays that can be used to determine whether the patient can go to surgery. As part of this testing, we can determine before surgery if there are residual effects of anticoagulants and antiplatelet agents. If the surgical procedure in a patient on one of the anticoagulants has a low risk for bleeding, anticoagulation may not have to be discontinued. If we’re talking about a tooth extraction, for example, as long as the International Normalized Ratio isn’t too high, the tooth can be removed. There are other minor procedures for which it’s simpler not to take the patient off the anticoagulant. If the procedure is a bit more invasive but the clotting risk is not that high, possibly for a patient who does not have inherited risk factors for thrombosis and his or her history of thrombosis is a weak one, you can advise simply discontinuing the warfarin for four to five days before surgery, without using a shorter acting anticoagulant as a ‘bridge’ between warfarin discontinuation and the start of surgery. Then you advise resuming the warfarin anticoagulation, knowing it might take about four to five days to return the patient to the full level of anticoagulation. Some patients, on the other hand, have had two or three clots previously. The reason for their anticoagulation is that they’re at high thrombotic risk. The physician stops the warfarin and initiates low-molecular-weight heparin or heparin. When the patient is close to surgery, then the LMW heparin or heparin is discontinued. This is known as ‘bridging’ therapy. The surgery is performed, and shortly after, the anticoagulation is restarted with heparin or low-molecular-weight heparin overlapping with warfarin, until warfarin is fully therapeutic. The aim here is to make a clot at the surgical site but at the same time not allow a thrombotic event to occur. Coumadin, which does not inhibit any coagulation factors, is different from all the other anticoagulants because the others are inhibitors. When you give them, they shut off the coagulation pathway. Coumadin does it differently: It reduces the synthesis of four of the factors—II, VII, IX, and X. When I was a young boy, I paid a lot of attention to cars, and when I was about eight years old my mother told me the factory that made the Studebaker was closing. I went outside to play, and the first car I saw coming down my street was a Studebaker. I went in to tell my mother she was wrong, because I had seen a Studebaker. She said, ‘No, Michael, there will still be Studebakers on the road. But because the factory is now closed, as the Studebakers break, they will be replaced with different cars, and it will take time for the Studebakers to go away.’ That’s what’s happening when you take Coumadin. Imagine factors II, VII, IX, and X being the Studebakers; they take a while to go away. Also, if a patient forgets his or her Coumadin one night, the patient will not be fully coagulable because it takes a while to regain the normal levels of factors II, VII, IX, and X. So the patient slides into anticoagulation, and if the drug is stopped, the patient slides back to coagulability. The International Normalized Ratio is used to monitor Coumadin therapy. Heparin, by comparison, inhibits the coagulation cascade at multiple sites. All of the activated coagulation factors, except factor III, are targets. Think of it as a shotgun. We monitor with the partial thromboplastin time (PTT) test. We can also use the assay for anti-factor Xa to assess the anticoagulant effect of heparin. When heparin is shortened and becomes low-molecular-weight heparin, the pharmacokinetic parameters of the drug change substantially. It inhibits the action of factors II and X, and the different low-molecular-weight heparins may inhibit II and X to different extents. Functionally, all of the low-molecular-weight heparins—Lovenox (enoxaparin), Fragmin (dalteparin), and Innohep (tinzaparin)—work about the same. We monitor with anti-factor Xa if needed. Some think that if monitoring does not have to occur on a regular basis, as with Coumadin, perhaps we don’t need to monitor at all. That is not true. Some patients who are supposed to take one pill take two by mistake, and some patients say, ‘I’m doing fine. I took my pill on Monday, and nothing’s feeling bad on Tuesday, so I’m skipping my pills on Tuesday and Wednesday.’ Such poor compliance is going to cause trouble for them. Patients who take too much can bleed, and those who don’t take enough can clot. We had better have an assay to find out what is going on. That is why, just because monitoring isn’t regular, it does not mean we don’t have to have laboratory tests to find out if these drugs are active and present in the right concentrations. Fondaparinux and rivaroxaban work not like a shotgun but like a rifle, directly inhibiting factor Xa, so the anti-factor Xa assay can be used to monitor if needed. Argatroban, lepirudin, dabigatran, and bivalirudin target IIa, the activated form of factor II, also known as thrombin. Argatroban and lepirudin are monitored with the PTT. Bivalirudin is such a hit to factor II, it’s so powerful, that it spikes the PTT to its maximum level. So we use the assay for activated clotting time, which is what you do when the PTT is going to be greater than 150 seconds and you want to get a sense of how much anticoagulant activity is present. Dabigatran is an inhibitor of thrombin, so if the patient is going to have surgery, we can use the thrombin time test to find out if there is residual drug. It is very sensitive to any residual dabigatran. Thus, if there is any question about a patient who is on dabigatran having surgery, one can do a thrombin time test. If the result is OK, the dabigatran is gone. Alternatively, a dilute thrombin time test can be used to better assess the actual anticoagulant effect in vivo. We monitor warfarin therapy today by using the International Normalized Ratio, or INR. We did not know long ago that as we were grinding up brains to do the prothrombin time (PT) test, there would be a difference in the assay if we ground up a human brain versus a rabbit brain to make thromboplastin for the PT assay. It made a lot of difference, and in fact, not all the rabbit brains had the same effect on the PT assay, either. They were not all prepared the same way. The INR equation is as follows: INR=[Patient PT/Mean of normal PT range]ISI.
Fig. 1 shows the impact of an ISI of 1.0 versus an ISI of 2.0. In this case of an INR of 3.0, if you have an ISI of 2.0, the PT value (the number above 10) is only a bit out—17.3. With the lower ISI of 1.0, the patient’s PT is 30 seconds. When the ISI is high, if only the PT is determined, the average clinician might say, ‘That’s only four seconds above normal; you’re taking more Coumadin. I’m changing you from 5 mg to 7.5 mg daily.’ There’s an important and unique aspect about Coumadin therapy. With heparin, if we want to prevent a clot, we give a small amount. If we want to treat a clot, we give a lot more heparin. For Coumadin, on the other hand, the target ranges are the same to prevent a clot and to stop an existing clot from getting larger. For a patient with atrial fibrillation taking warfarin to prevent a stroke, for example, the target INR is 2.0 to 3.0. It’s the same target for a patient who experiences a deep vein thrombosis or pulmonary embolism. That presents a problem because there are a lot of people with atrial fibrillation, and we have to get them into a target range that’s pretty high. That means it’s a lot easier to get bleeders with Coumadin because the target range for prophylaxis is the same as it is for many of the treatments. And for some conditions, the treatment target is 2.5 to 3.5. Mechanical heart valves, for example—using warfarin to prevent a clot has an INR target of 2.5 to 3.5. When do we start running into trouble? When you get down to an INR of about 1.5, you’re in the zone of three to five times the risk of stroke for the patient with atrial fibrillation versus when the patient is in the therapeutic range. You can tolerate the 1.9 and 1.8 for a while, but it’s much better to be at 2.0–3.0. If we anticoagulate too much, the patient could hemorrhage inside the brain—subdural, subarachnoid, or even worse, intracerebral. How high above the 2.0–3.0 range do you start to worry? 3.7 to 4.3 does not look too bad. But when you start to approach an INR of 5.0, the risk for an intracranial hemorrhage is now four- to fivefold above what the patient would have otherwise. It is a narrow therapeutic INR window. There’s a lot of biological, patient-to-patient variability no matter how compulsive the patient is about taking Coumadin. Many advocate the use of pharmacogenomics because up to 30 to 40 percent of the impact of warfarin is based on the enzymes that affect the metabolism of warfarin: vitamin K epoxide reductase and cytochrome 2C9. And it is true that some patients should be started not on a 5-mg daily dose, but instead 2.5, and pharmacogenomics may be of help. But there is a problem. We give Coumadin and then we monitor with the INR. Let’s say it takes six days to get the results of the pharmacogenomic studies. By that time, you’ve probably figured out the patient needs a lower dose of Coumadin, because the INR was too high. For most cases, you are already into the therapeutic INR range about the time you get the pharmacogenomic results. So the reaction to the use of pharmacogenomics for warfarin has been mixed, and there are data to suggest that the performance of pharmacogenomic studies for warfarin do not change outcome.
In the algorithm for warfarin use (Fig. 2), the box on the left says to select the target range, 2.0 to 3.0 or 2.5 to 3.5. To its right is ‘overdose,’ and if that’s ‘no,’ you’re winning the game. On the other hand, if it is ‘yes,’ you have to know how to reverse the warfarin effect. Step one if you think the patient is going to bleed to death: Stop the warfarin and give vitamin K. Some people advocate that it be given intravenously; we worry about anaphylaxis with that. It can also be delivered sub-Q, and oral vitamin K reverses the warfarin more slowly. Plasma is given to replace those four missing factors; it’s like pouring in Studebakers, to use the earlier analogy. Some advocate the use of prothrombin complex concentrates. In Europe they have prothrombin complex concentrates that have all four of the missing factors. Most of the ones we use are low in one of them—factor VII. But prothrombin complex concentrates can also be used. Next, am I worried about regaining the anticoagulated state as soon as possible? If ‘yes,’ and if the INR is, say, less than 10 without bleeding, you have a couple of choices. You could tell the patient to not take warfarin that night and the next night, and let the INR decline toward the therapeutic range. Alternatively, if the INR is above 10 and other risk factors for bleeding exist, you might want to stop the warfarin and give vitamin K—in this case, one might give oral vitamin K, 1.0–2.5 mg. If the INR returns to that target range, you often decrease the Coumadin dose, and the patient is back where he or she needs to be. To sum up, the three questions about reversal are whether the patient is going to bleed to death, whether you can trickle the patient back by discontinuing Coumadin, or whether you have to add vitamin K. Who would have thought heparin is sugars?
It is glucosamine and uronic acids in alternating sequences, and these sugars have attached sulfate groups. Heparin is a very negatively charged compound, and it’s purified from pig intestines. What you get from the pig intestines are long chains, some that are a little shorter, and others that are tiny. You can cut the big ones into pieces, enzymatically or chemically, and that’s one way to make low-molecular-weight heparin. In Fig. 3 it is drawn as a staircase on the left side of each of the four panels. Negative charges are in the white ovals. Heparin works for anticoagulation through a protein called antithrombin, or AT. The negative charges on heparin bind the positively charged portion of antithrombin. When antithrombin is activated and becomes a much better anticoagulant, you will see a change in its conformation, and this change allows it to bind to a coagulation factor and inhibit it. If the heparin is long enough, the heparin will, as I call it, step on the foot of the coagulation factor, while the antithrombin punches it in the head. This is the way that full-dose unfractionated heparin, with sufficiently long chains, can inactivate all of those activated coagulation factors. The shorter low-molecular-weight heparin still binds to antithrombin, but it is not touching the coagulation factor. Factor Xa does not have to have ‘its foot stepped on’ by low-molecular-weight heparin to be inactivated. That is why low-molecular-weight heparin largely inhibits Xa. Inhibition of IIa activity is because not all of the big pieces of heparin are chopped into little pieces, so you can inhibit IIa when you make a LMW heparin. Heparin works right away; remember the Studebaker story about Coumadin. If you have a two-inch clot by your ankle, you do not want to wait four days to get anticoagulated. For patients with active clot formation, when we give warfarin, we also give a drug like heparin or LMW heparin that works right away, and overlap it with warfarin. Generally, the overlap is five to nine days until you have two INRs where you want them to be, showing Coumadin is therapeutic, and then other anticoagulants can be discontinued.
Fig. 4 is the algorithm for heparin use, including LMW heparin. For a small amount of heparin given as prophylaxis, there is no need to monitor. In an adult who is not obese or too thin and who has normal renal function, there’s no monitoring for LMW heparin. It’s full-dose unfractionated heparin that needs regular monitoring. The PTT you want to get to is at least two times the mean of the normal range. Let’s say that it is 26 to 34 seconds, so the mean is 30 and two times that is about 60 seconds. If the patient is not overdosed, then maintain that heparin dose. If you use anti-factor Xa monitoring for unfractionated heparin, the target range is 0.3–0.7 IU/mL. But what if bleeding occurs in a patient on heparin? Do not use the same strategy used for warfarin. Here you have to use a compound that is positively charged—protamine sulfate. This protamine will bind to the heparin and neutralize it. You may have to do it twice to stop any kind of bleeding with an overdose of heparin. On the other hand, if you do not think the patient is going to bleed to death, you can wait, discontinue the heparin, and watch carefully. Unfractionated heparin has a half-life of about an hour. After two hours, you’ve eliminated about 75 percent of it, so often you do not have to do anything. So the big question is: How worrisome is the bleeding, and should an attempt be made to inhibit with protamine sulfate? For LMW heparin, Lovenox (enoxaparin) and Fragmin (dalteparin) make up virtually all of the market. I refer to them interchangeably as low-molecular-weight heparins. How do we monitor these compounds in the laboratory? It is renal function we are first concerned about. The estimated glomerular filtration rate is a good assay to use. By some assessment of renal function, however, you need to know whether the dose of LMW heparin should be reduced. Interestingly, people who are too big or too small do not metabolize low-molecular-weight heparin as expected. Women who are pregnant and even the babies, especially the neonates, metabolize it faster, so there are dosage adjustments to be made, and that’s why we use anti-factor Xa measurements for LMW heparin. I’ve had patients who have been on LMW heparin for years, and about once a year, I measure their anti-factor Xa. The target range for LMW heparin is 0.5 to 1.0 IU/mL. Here is a common mistake. In fact, one of my former patients, in Massachusetts, called me in Tennessee, and she’s had pelvic bleeding—terrible compression, pain. I knew she was on LMW heparin, and I said, ‘Please tell your doctor to get an anti-factor Xa assay and to send me the result so we can be sure you don’t need a dosage adjustment.’ What they did was to order the factor X, not the anti-factor Xa. When we’re looking at factor X, we’re trying to find out if the patient is deficient in the factor, and on that basis, predisposed to bleed. That is a different test from anti-factor Xa, which is used to monitor an anticoagulant effect. You can reverse low-molecular-weight heparin (protamine sulfate, one percent solution), though maybe not all of the anti-Xa activity. But even if you reverse a significant portion of it, it should stop the bleeding in most cases. |