Validating and verifying molecular tests in cytopathology

| Validating and verifying molecular tests in cytopathology |
|
|
|
May 2011 Lynnette Savaloja, SCT(ASCP) This article provides an overview of what is required and recommended to properly verify or validate, analytically and clinically, new molecular tests to be performed in the cytopathology laboratory. FDA clearance designations It is important to understand the Food and Drug Administration clearance designations for laboratory tests and reagents before implementation. These designations will determine the type of validation or verification that must be performed. For the purposes of this article, three clearance designations will be reviewed: in vitro diagnostic products (IVD), analyte specific reagents (ASR) (including FDA-modified tests), and research use only (RUO).1 In vitro diagnostic products. In regard to laboratory testing, IVDs are reagents or instruments intended for use in diagnosing disease or other conditions in patients. The FDA clears or approves these products for one or more specific intended uses with established analytical and clinical performance characteristics. IVD products are designated from class I to class III, with class III encompassing most laboratory tests. Class I includes items such as bandages and handheld surgical instruments; class II includes items such as powered wheelchairs, infusion pumps, and surgical drapes.2 There are two subcategories for class III products: premarket approval (PMA) and 510(k). The PMA is the most stringent regulatory class and requires a process of scientific review to ensure the safety and effectiveness of class III products. It is through this submission process that the analytical and clinical performance characteristics are proven and approved by the FDA. It is this PMA designation for which the term “FDA approved” is used. A 510(k) is a premarket submission made to the FDA to demonstrate that a product is at least as safe and effective as a product that has already gone through a PMA submission and is currently on the market. A 510(k) product is “FDA cleared.” Analyte specific reagents. A more recent FDA designation is the analyte specific reagent. The FDA developed this designation in 1997 to regulate and ensure the quality of homebrew laboratory-developed tests (LDTs). The ASR is an active ingredient of an in-house test. It is not a test in and of itself, nor can it be marketed as part of a test kit. In contrast to an IVD, the FDA has not validated or approved any intended use or performance characteristics for this type of product. It is the responsibility of the laboratory performing the test to do so. In addition, the FDA requires users of ASR products to include a disclaimer on all patient results obtained using ASR products indicating that the performing laboratory has validated the test’s perŁformance. Although not a true FDA classification, the FDA-modified test must be included in this review. “FDA-modified” is the term used when an FDA-approved or -cleared test is used in a manner that does not follow the FDA approved use of the product as specified in the package insert. This is considered off-label use and must be validated in the same manner as an ASR. The FDA published a guidance document on ASR use in September 2007 that went into effect in September 2008. The purpose of this document was to clarify the ASR rules and introduce more stringent enforcement in this area.3 Research use only. Research-use-only assays are intended for research purposes and generally are not recommended for clinical testing. However, the CAP has made a provision for them in its laboratory accreditation checklist. The CAP says that RUOs may be used in laboratory-developed tests only if the laboratory has been unable to find IVD- or ASR-class reagents for that test. The laboratory director is responsible for the documentation of this search. Use of assays with this designation may not be reimbursable. Validation versus verification The terms verification and validation are not synonymous. This is especially true when discussing the performance of laboratory tests. Unfortunately, some accreditation agency standards use these terms inconsistently and interchangeably, which can cause confusion before new tests are implemented in the laboratory. Understanding the distinction between validation and verification is important in optimizing patient care and laboratory operational efficiency, as well as in fulfilling government regulatory requirements. Verification: ‘Did I do the thing right?’ Verification is the process by which the lab verifies that the established performance claims of an IVD test or product can be replicated in the lab before patient testing. In essence, verification establishes that the laboratory performing the test executes the test procedures correctly and ensures that instrumentation and reagents work properly. Verification is acceptable in circumstances in which the test is performed and used in the manner directed in the package insert. Any other off-label use would require validation by the performing laboratory. The ASCO/CAP HER2 testing guidelines published in 20074 and the ER/PR guidelines published in 20105 are in part a result of insufficiently stringent verification of the performance of an IVD in many laboratories, resulting in excessive interobserver variability of test results. Validation: ‘Did I do the right thing?’ Validation is the process by which the laboratory measures the clinical efficacy of the test in question by determining its performance characteristics when used as intended. This is necessary to prove that it performs as expected and achieves the intended result. Validation is required when using laboratory-developed tests, ASR products, or FDA-modified tests. Regulatory requirements for performance Just as it is important to understand the difference between validation and verification, it is also important to be aware of the accreditation requirements for test performance. CLIA. CLIA addresses test performance in standard 493.1253: Establishment and verification of performance specifications:6 (b)(1) Verification of performance specifications. Each laboratory that introduces an unmodified, FDA-cleared or approved test system must do the following before reporting patient test results: (i) Demonstrate that it can obtain performance specifications comparable to those established by the manufacturer for the following performance characteristics: (A) Accuracy Note that this standard uses the word “verification” and the phrase “unmodified, FDA-cleared or approved test.” This describes an IVD product. Further in the standard is the following requirement: (b)(2) Each laboratory that modifies an FDA-cleared or approved test system, or introduces a test system not subject to FDA clearance or approval (including methods developed in-houseģ), or uses a test system in which performance specifications are not provided by the manufacturer must, before reporting patient test results, establish for each test system the performance specifications for the following performance characteristics:
(i) Accuracy Again, it is important to be aware of the context of this standard. The phrase “system not subject to FDA clearance” describes a test developed with ASRs, as well as FDA-modified tests. These are the requirements for performing a test validation as described by CLIA. An example of the need for validation of a system not subject to FDA clearance is immunocytochemistry when performed on cytologic smears instead of formalin-fixed paraffin-embedded tissue (FFPE). Most if not all FDA-approved immunohistochemistry kits for clinical use were approved based on protocols using FFPE tissue. It cannot be assumed that the staining of tissue prepared or preserved in a manner other than FFPE, such as with ethanol fixation, will match that of FFPE tissue. Therefore, the performance of the assay must be validated if the tissue preparation differs from that specified in the FDA-cleared or -approved protocol. A validation protocol for this situation would involve collecting a group of FFPE cases with the diagnoses of interest including relevant differential diagnoses and cases that one would expect to be unequivocally negative, and corresponding cytology preparations from the same cases that may have been prepared from prior FNAs or touch or scrape preparations. The immunochemical stains would then be performed on the smears and FFPE tissue, and the proportion of concordant and discordant cases tabulated and subjected to statistical analysis. While no definitive protocol for statistical analysis of data collected for validation studies has been established, a kappa analysis with confidence intervals might be appropriate for this situation. Formal consultation with a statistician may be helpful in ensuring a robust validation protocol. Joint Commission. The Joint Commission accreditation standards are similar to the CLIA standards, but the terminology is different.7 Standard 02.01.01 from the Quality System Assessment chapter of the 2011 Joint Commission Laboratory Accreditation Standards manual requires that “the laboratory verifies tests, methods, and instruments in order to establish quality control procedures.” This standard also applies to instruments on loan when the original instrument is under repair. The following Elements of Performance (EP) from this standard further clarify the Joint Commission validation/verification requirements: EP 1: When adding or replacing an unmodified FDA approved test, the laboratory verifies the manufacturer’s performance specifications, including:
EP 2: When adding or replacing a modified test—defined as FDA approved tests that have been modified from the package insert, tests developed in the laboratory with no FDA evaluation or tests not subject to FDA clearance—the laboratory must establish written performance specifications that include:
Although not specifically stated in this Element of Performance, the establishment of performance characteristics as described above is a validation process. This standard continues: EP 3: When replacing an old test, method or instrument, the laboratory’s verification includes a correlation between the old test, method or instrument and new one, and the correlation is documented. EP 4: For a new test, method, or instrument, the laboratory verifies that the reference intervals (normal ranges) apply to the test, method, or instrument and population served. The verification is documented. EP 5: The laboratory performs verifications for each new test, method, or instrument prior to reporting patient results. These verifications are documented. EP 6: The laboratory’s verification includes the establishment of written quality control procedures for each testing system or methodology. The Joint Commission includes additional validation requirements for molecular testing. Standard 15.02.01 requires the laboratory’s verification studies for molecular testing include representatives from each specimen type expected to be tested in the assay and specimens representing the scope of reportable results. The EPs for this standard are as follows: EP 1: The laboratory’s verification studies for molecular testing include positive and negative representatives from each specimen type expected to be tested in the assay. EP 2: The laboratory’s verification studies for molecular testing include specimens representing the scope of reportable results. EP 3: The laboratory performs verification studies for molecular testing. The verification studies are documented. Note that the Joint Commission uses the term “verification” for this standard and corresponding EPs. A verification process would be used if an FDA-cleared or -approved molecular test is implemented. However, if the test is developed by the laboratory (an LDT) or if it is an FDA-modified molecular test, a validation study would be required. College of American Pathologists. There are three CAP checklists in which we find standards related to test verification and validation.8 The CAP general checklist says the following: GEN.42020: Has the laboratory verified or established and documented analytic accuracy and precision for each test? GEN.42025: Has the laboratory verified or established and documented the analytic sensitivity (lower detection limit) of each assay, as applicable? GEN.42030: Has the laboratory verified or established and documented analytic interferences for each test? Note that the CAP checklist uses the terms “verified or established.” The word “established” here refers to test validation. The CAP cytopathology checklist has the following standards: CYP.05257: Is there documentation of adherence to the manufacturer’s recommended protocol(s) for implementation and validation of new instruments? Most instruments will have an IVD classification, so we can assume that the CAP is referring to verification that the instrument works before we use it to test our patients. Finally, the CAP molecular checklist says: MOL.30957: For FDA-cleared/approved assays, are performance criteria verified by the laboratory? MOL.31590: Is the clinical performance characteristics of each assay documented, using either literature citations or a summary of internal study results? Up to this point, we have been describing analytical performance. This is different from clinical performance. The two are different and will be discussed in the next section. However, it is important to note that the CAP has included an educational comment regarding clinical performance in this checklist that recognized that, in contrast to analytic validity, establishing clinical validity may require extended studies and monitoring that go beyond the purview or control of the individual laboratory. In such cases, it is acceptable to provide documentation in the form of peer-reviewed studies in the scientific literature. It is essential that directors use their highest professional judgment in evaluating the results of such studies and in monitoring the state-of-the-art worldwide as it applies to newly discovered gene targets and potential new tests, especially those of a predictive or incompletely penetrant nature. Analytical versus clinical performance characteristics Analytical performance measures the results of a test in comparison with a reference test or gold standard. Clinical performance refers not only to the results of the test but also to how those results correlate with a patient’s disease state and clinical outcome. This difference as it applies to human papillomavirus testing was addressed in two articles.9,10 These authors stress that it is important not only that the test be analytically validated but also accurate in judging whether a clinically relevant HPV infection is present. To be clinically relevant, the test must have the ability to predict the actual presence of the disease (now or for a defined period in the future). To validate this type of performance characteristic, patients must be biopsied and followed for the presence or progression of cervical disease. Elements of Performance The following section describes the parameters used to measure the performance characteristics of diagnostic tests and assess how well tests meet user expectations for diagnostic utility.11¢13 Accuracy and precision. Accuracy measures the closeness of the agreement between a test result and the true value or result. In other words, if the test is positive, does the patient really have the condition? Precision, on the other hand, measures the closeness of repeated tests to the same value or result. Or, is the same result achieved every time the same sample is tested? All variables of precision must be measured during the validation or verification of a laboratory test. These precision studies include “between run” (will the test have the same result for the sample on different runs on different days?), “within run” (will the test have the same result for the same sample tested multiple times in the same run?), and “between user” (does the result for this sample change with different personnel performing the test?). Sensitivity and specificity. Sensitivity and specificity are among the most commonly discussed measures of performance. Assessing these two parameters requires determining the number of true positive (TP), true negative (TN), false-positive (FP), and false-negative (FN) results in a validation data set. These terms are defined as follows: True positive (TP): A positive test result for a patient in whom the condition or disease is present. True negative (TN): A negative test result for a patient in whom the condition or disease is absent. False-positive (FP): A positive test result for a patient in whom the condition or disease is absent. False-negative (FN): A negative test result for a patient in whom the condition or disease is present. Sensitivity is the probability that a test is positive when the disease/condition is present. Mathematically, it is: TP TP + FN Specificity is the probability that a test is negative when the disease/condition is absent. Mathematically, it is: TN TN + FP A related concept that is most commonly associated with analysis of rescreening data is the false-negative fraction. This is the ratio of FN cases divided by all positives, or: FN TP + FN The false-negative fraction is not FN divided by all negatives: FN TN + FN Positive and negative predictive value. Positive and negative predictive values are important parameters in assessing the clinical impact of tests. Though they are related to sensitivity and specificity, they are different and not interchangeable. Positive predictive value is the probability that disease is present when the test is positive. Mathematically, it is: TP TP + FP Negative predictive value is the probability that disease is absent when the test is negative. Mathematically, it is: TN TN + FN An important characteristic of positive and negative predictive values is that they vary depending on the prevalence of the disease being tested for. In contrast, sensitivity and specificity are characteristics of the test itself and do not vary with the prevalence of disease. Kappa. Cohen’s kappa statistic, often referred to as simply kappa, is a measure of interobserver agreement. It may be applied when two tests that yield categorical or ordinal data are being compared and the data can be compiled in an N ? N table. The kappa statistic is designed to measure the agreement that is in excess of that which would be expected to occur randomly when two observers are compared. Kappa scores range from -1 to 1, with 1 indicating perfect agreement, 0 indicating no agreement, and negative scores indicating inverse agreement. Negative scores are usually not seen because presumably there is at least some degree of agreement between the two tests being compared. The level of agreement indicated by kappa scores has been described as seen in the following table:
If the data are ordinal, the weighted kappa statistic can be used. Categories are ordinal if they can be ranked, such as in the HER2 scoring system. With the weighted kappa statistic, the more discrepant pairs of observations contribute less to the total score, and the more concordant pairs of observations contribute more. The computational details of kappa are beyond the scope of this article. Details are available in Viera.14 Many statistical software packages can calculate kappa (MedCalc, SPSS, SAS, VassarStats site); Web-based calculators are also available.15 Receiver operating characteristic. The receiver operating characteristic (ROC) may be useful in the clinical validation of a test whose outcome variable is binary, for example disease present or disease absent. The measured parameter can be an interval or ordinal variable. The ROC is a graph that plots the TP rate as a function of the FP rate (1 ¢ specificity) as the cutoff point is varied. Each point on the curve represents a sensitivity/specificity pair that corresponds to a particular cutoff point. An example of a curve representing various cutoff points between the two outcome states is plotted on this graph. 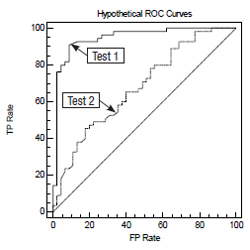
This is a graph with two ROC curves representing test 1 and test 2. The ability of the test to discriminate between the two outcomes is proportional to the area under the curve. More specifically, the area under the curve is equal to the probability that two randomly selected samples, one from each of the two outcome groups, will be ranked correctly. Therefore, test 1 is a better test than test 2 in this example. Additional information on the use and interpretation of ROC curves can be found in Hanley and McNeil,16 and Swets.17 Software that can prepare ROC curves, compute the area under the curve, and compare two ROCs is available on the Web and as standalone packages (SPSS, MedCalc, Vassar statistics, U of Chicago, many others). Confidence intervals. Attention must be given to confidence intervals of statistical parameters that are used in validation studies. The upper and lower 95 percent confidence intervals should fall within an acceptable range (similar to that of the reference test) for the clinical situation in which the test will be used. The statistics software mentioned in this article will compute confidence intervals. The details of computing confidence intervals are beyond the scope of this article. Summary As laboratory testing becomes more complex and LDTs proliferate, proper understanding and application of test verification and validation are of the utmost importance for patient safety and optimal clinical outcomes.18 It is the intent of this article to present an introduction to the pertinent parameters and regulations that must be understood to carry out this responsibility. Acknowledgements This document is promulgated by the Cytopathology Education and Technology Consortium (CETC) to assist the cytopathology community in understanding the concepts of validation and verification and in meeting pertinent regulatory requirements, and to promote superior clinical laboratory practice in cytopathology. The CETC is composed of representatives from the following professional societies with interest in cytopathology: American Society for Clinical Pathology, American Society of Cytopathology, American Society for Cytotechnology, College of American Pathologists, International Academy of Cytology, and Papanicolaou Society of Cytology. The current representatives are as follows: George Birdsong, MD, co-chair, Emory University School of Medicine, Atlanta; Lynnette Savaloja, SCT(ASCP), co-chair, Regions Hospital, St. Paul, Minn.; Karen Atkison, MPA, CT(ASCP), TriPath Imaging, Inc., Burlington, NC; Guliz A. Barkan, MD, Loyola University Medical Center, Maywood, Ill.; Diane D. Davey, MD, University of Central Florida College of Medicine, Orlando; Ritu Nayar, MD, Northwestern Memorial Hospital, Chicago; Martha B. Pitman, MD, Massachusetts General Hospital, Boston; Celeste N. Powers, MD, PhD, Medical College of Virginia, Richmond; James M. Riding, BS, CT(ASCP), ARUP Laboratories, Salt Lake City; Patricia G. Wasserman, MD, Hofstra North Shore LIJ School of Medicine, Lake Success, NY. The CETC gratefully acknowledges Elizabeth Jenkins, executive director of the American Society of Cytopathology, for her invaluable assistance and support to the CETC and in the preparation of this manuscript. References |
