 CAP TODAY Pathology/Laboratory Medicine/Laboratory Management
CAP TODAY Pathology/Laboratory Medicine/Laboratory Management

William Check, PhD
January 2017—Detecting pathogenic organisms with PCR has become a staple of the clinical microbiology laboratory, so much so that it seems like it has always been there. A more advanced molecular technique—unbiased metagenomic next-generation sequencing—will increasingly become a part of infectious disease diagnosis because it has several advantages over PCR. While it will be demanding to perform at first, it, too, may become a standard method in the clinical microbiology laboratory.
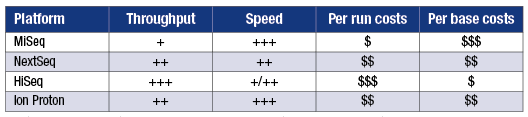
Whether samples need to be run individually or can be batched influences costs and workflow. Compiled by Robert Schlaberg, MD, MPH.
“In contrast to most of our current tests, you can use a metagenomic approach to find any and all potential pathogens in a patient sample without having to know what you’re looking for,” says Robert Schlaberg, MD, MPH, an assistant professor of pathology at the University of Utah and medical director of infectious diseases at ARUP Laboratories. While PCR can be fast and effective, it needs to be targeted. “So it is difficult to use when a condition can be caused by many microbes and pathogens. In that situation, you need to use a battery of tests, which can become lengthy and expensive and often doesn’t yield a result.” Such is the case for pneumonia, sepsis, encephalitis, meningitis, and diarrhea.
“The advantage of a metagenomic approach is that you can start without having a hypothesis,” says Dr. Schlaberg, who spoke with CAP TODAY and presented in November at the Association for Molecular Pathology meeting on universal pathogen detection directly from specimens in the diagnostic laboratory. Another advantage of metagenomics is that it can detect bacteria, viruses, fungi, and parasites in one assay.
“In our lab, the process from sample to report takes about 1.5 days,” Dr. Schlaberg says of the complex workflow. “It takes a full shift at least to process the sample; sequencing takes overnight.” Still, he can envision a simpler workflow and a future in which metagenomic sequencing replaces many current tests. “Right now to get started is challenging,” he says, but it’s easy to forget that PCR was also once labor-intensive and demanding to run. “It will be the same as with PCR,” Dr. Schlaberg says. “Technology will evolve, and we will get to a point where metagenomic sequencing is done in most large labs.”
Dr. Schlaberg and colleague Mark Yandell, PhD, a professor of human genetics at the University of Utah School of Medicine and co-director of the USTAR Center for Genetic Discovery, have made a start toward the future by co-developing a semiautomated informatics package for analysis of metagenomic sequence data as a clinical microbiology detection tool. Their program, called TaxonomerDx, will be deployed in a few months at ARUP Laboratories. Its initial indication will be for pneumonia.
Unexplained infectious illness ranges from 40 percent upward for sepsis, infectious diarrhea, pneumonia, and febrile neutropenia. CNS infections top the list with an 80 percent “unknown” rate.
“Even for [CNS] cases where we know what causes an infection, we need a very sensitive test to detect an organism,” Dr. Schlaberg says. “Often with viral infections they are there for only a few days, so we could be collecting a sample after the organism has gone.” Then, too, noninfectious conditions can look similar to a CNS infection. Dr. Schlaberg cites post-infectious encephalitis and autoimmune conditions as examples of conditions that may clinically look like an infection. Often when a patient presents to the hospital with a suspected infection, he or she is treated quickly, even before optimal specimens are collected. “So the antibiotics can be in the specimen and inhibit growth of the organisms in the lab.” Many of these obstacles can be overcome by a metagenomic technique, like the Taxonomer-based technology platform the University of Utah has developed, Dr. Schlaberg says.
Unbiased metagenomic sequencing means all the nucleic acid in the sample is submitted for sequencing and analysis. “Unbiased” means all the nucleic acid is analyzed just as it is in the specimen, without being amplified.
In the metagenomic technique, as in all next-generation sequencing, sensitivity depends on the number of times each nucleic acid segment is sequenced, or read (“read depth”). External and internal controls are essential. “Many routine kits and reagents used in NGS processes are contaminated with microbial DNA and RNA,” Dr. Schlaberg warns. “With unbiased metagenomics sequencing, we routinely detect these microbial nucleic acids. Using the right negative controls helps identify these as contaminants. This sounds trivial, but the importance is hard to overstate.” He shares a case in which a novel virus found in clinical samples turned out to originate from contaminated silica-binding spin columns used for nucleic acid extraction (Naccache SN, et al. J Virol. 2013;87[22]:11966–11977).
Selecting an instrument for metagenomic sequencing involves tradeoffs among throughput, turnaround time, and cost. Dr. Schlaberg has tabulated relative throughput and speed for several instruments and both per run and per base costs (see box, this page). “In infectious disease diagnosis you need to be fast,” he notes. “We use the Illumina NextSeq platform. Of the available instruments, it gives us the best combination of rapid turnaround time, per base sequencing cost, and per run cost.”
This is just a start, though, he says, adding, “We are always looking for ways to speed up laboratory protocols and sequencing times.”
Still needed, he says, are user-friendly data analysis solutions for the diagnostic setting. Ideally they should require minimal bioinformatics expertise and be easily updatable and expandable. All current databases are imperfect and incomplete and contain errors, he cautions, and they require curation. “Informatics for metagenomic application used to be very slow.”
“Until a couple of years ago, alignment-based methods were used for data analysis,” Dr. Schlaberg explains. Because every one of millions of sequences from the patient sample needs to be compared to millions of reference sequences, these are computationally intense analyses and took days or weeks to complete. Advances in computer science made possible alignment-free analysis methods, which can be completed in minutes.