CAP TODAY and the Association for Molecular Pathology have teamed up to bring molecular case reports to CAP TODAY readers. AMP members write the reports using clinical cases from their own practices that show molecular testing’s important role in diagnosis, prognosis, and treatment. The following report comes from Emory University School of Medicine. If you would like to submit a case report, please send an email to the AMP at amp@amp.org. For more information about the AMP and all previously published case reports, visit www.amp.org.
Julia An, MD
Debra Saxe, PhD
Jaime Vengoechea, MD
Shiyong Li, MD, PhD
July 2023—Acute promyelocytic leukemia (APL) constitutes approximately five to eight percent of cases of acute myeloid leukemia (AML) and commonly presents in young adults.1 Prompt diagnosis is essential because of the frequent association with life-threatening disseminated intravascular coagulation (DIC) as well as the availability of therapeutic treatment options such as all-trans retinoic acid (ATRA) and arsenic trioxide (ATO). Two morphological variants of APL have been described: hypergranular (typical) and hypogranular (microgranular).
The hypergranular variant is associated with leukopenia and frequent abnormal promyelocytes with abundant cytoplasmic granules and bundles of Auer rods. Immunophenotypically, the abnormal promyelocytes demonstrate increased side scatter with low to absent expression of HLA-DR and CD34, as well as leukocyte integrins (CD11a, CD11b, and CD18). Variable CD13, uniform CD33, and strong cytoplasmic myeloperoxidase (MPO) expression are also characteristic features.1
On the other hand, the hypogranular variant is associated with leukocytosis and displays morphologic features of abnormal promyelocytes that have sparse or fine granulation and irregular bilobed or butterfly-shaped nuclei.1 Immunophenotypically, the abnormal promyelocytes demonstrate expression of CD13, CD33, and MPO with dim expression of HLA-DR and CD34 as well as aberrant expression of CD2 and/or CD56. CD2 expression has been associated with FLT3 mutations of which internal tandem duplications (ITD) make up the majority.2
GATA2 is located on chromosome 3q21.2 and encodes a zinc finger transcription factor, GATA2.3 GATA2 is critical in the formation and maintenance of hematopoietic stem cells as well as the production of megakaryocytes, mast cells, natural killer (NK) cells, and monocytes. De novo sporadic germline mutations or autosomal dominant inheritance of a mutated copy of GATA2 may lead to GATA2 deficiency.3 GATA2 deficiency is a complex multisystem disorder that may manifest with bone marrow failure, hematologic malignancies, and severe immunodeficiency.4 Pathogenic germline variants in GATA2 have been associated with a variety of clinical symptoms that have been classified into various groups such as MonoMAC (monocytopenia and mycobacterial infection), DCML deficiency (dendritic cell, monocyte, B and NK lymphoid), and Emberger syndrome.3 Here we describe a germline GATA2 variant in a patient with APL, which demonstrates an unusual morphology and immunophenotypic profile.
Case. A 21-year-old adopted male patient with a past medical history of autism spectrum disorder and obsessive-compulsive disorder presented to an urgent care center with nausea, vomiting, congestion, mild cough, and abdominal pain in August 2021. A complete blood count demonstrated hemoglobin of 11.7 g/dL (11.4–17.1 g/dL), white blood cell count of 12.4 × 103/µL (3.1–10 × 103/µL), and platelet count of 41.0 × 103/µL (126–362 × 103/µL). In addition, the WBC differential count revealed increased blasts (29 percent). Blood coagulation tests were significant for prothrombin time of 19.1 seconds (11.9–14.1 seconds) and INR of 1.66 (0.9–1.1). Repeat blood cultures demonstrated no growth, and testing for COVID-19, HIV, and hepatitis C viruses was negative. CT imaging of abdomen/pelvis revealed splenomegaly with findings suggestive of splenic infarct with consideration for lymphoma or other lymphoproliferative process. He was subsequently transferred to Emory University Hospital for further evaluation of suspected acute leukemia.
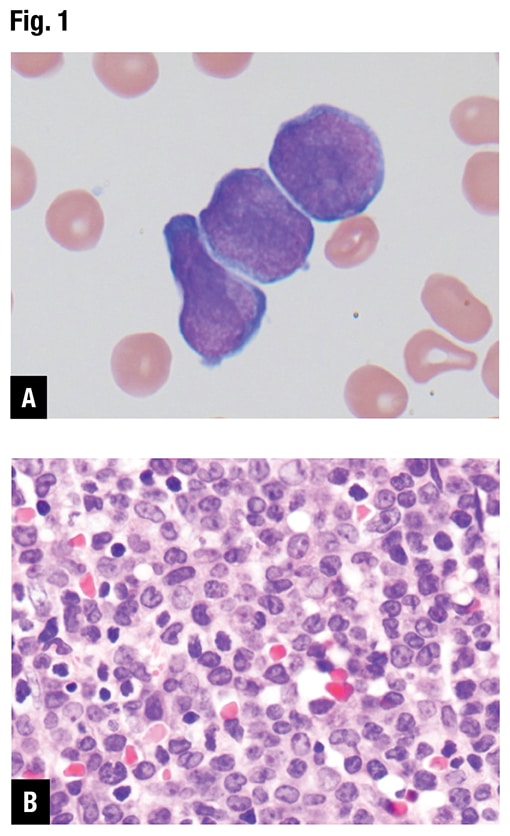
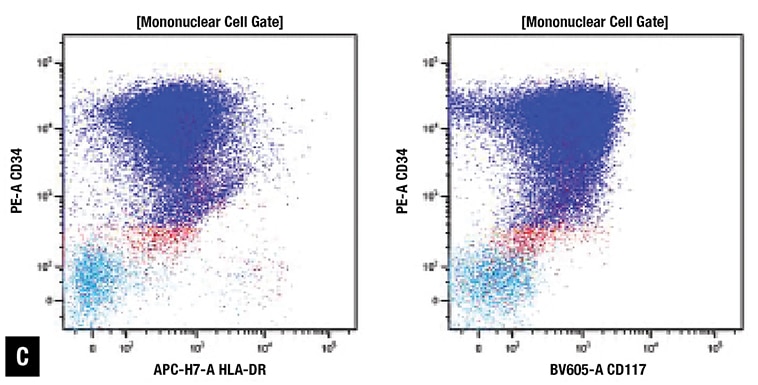
At Emory, peripheral blood smear review revealed approximately 70 percent blasts with scant basophilic cytoplasm and irregular nuclear contours (Fig. 1A). No definite Auer rods were identified, and neither classical abnormal promyelocytes nor their bilobed variants were evident. Bone marrow biopsy showed a hypercellular marrow with sheets of immature mononuclear cells with irregular nuclei and scant to moderate amount of cytoplasm (Fig. 1B). Flow cytometric immunophenotyping showed 86 percent of analyzed cells to express CD2, CD13, CD33, partial CD36, CD64, CD117, CD123, CD200, and CD45 (dim) with coexpression of bright CD34 and moderate HLA-DR. Representative flow cytometry dot plots are shown in Fig. 1C.
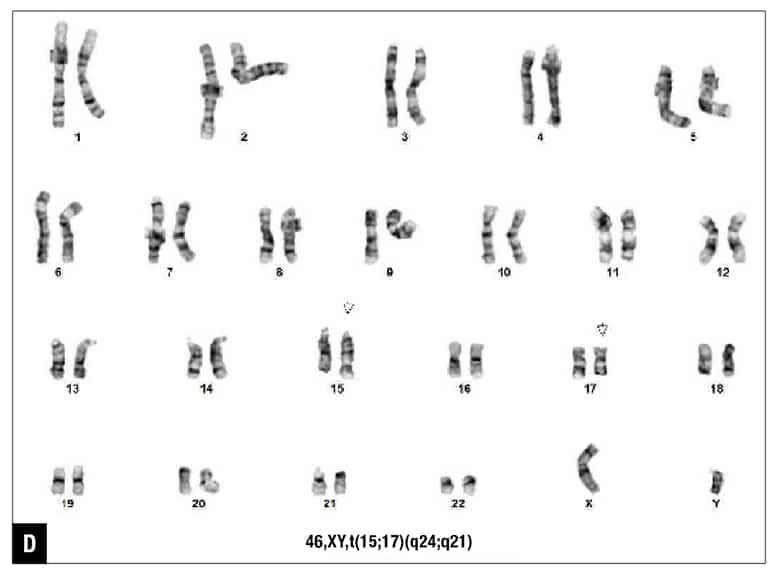
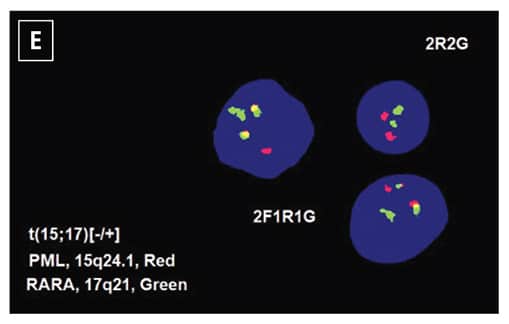
The patient was diagnosed with acute myeloid leukemia, and 7+3 chemotherapy regimen (cytarabine and daunorubicin) was initiated. Five days after the initiation of chemotherapy, routine chromosome analysis demonstrated an abnormal male karyotype with translocation between the long arms of chromosomes 15 and 17, t(15;17)(q24;q21), in 18 of 21 metaphases examined (Fig. 1D). Also, FISH confirmed the presence of t(15;17)(PML::RARA) in 82 percent of the interphase nuclei (Fig. 1E). Taken together with the morphologic and flow cytometry findings, the diagnosis was revised to APL following the results of ancillary studies,5 and ATRA therapy was initiated.
Next-generation sequencing was performed at Emory University Hospital using the in-house targeted myeloid mutation panel, which simultaneously detects single nucleotide changes, insertions and deletions, and internal tandem duplications of 75 genes involved in myeloid neoplasms (e.g. ABL1, FLT3, ASXL1, GATA2, etc.). Briefly, genomic DNA was isolated from bone marrow samples using a Qiagen DNA preparation kit. The targeted exons of the 75 genes were amplified using Anchored Multiplex PCR (AMP). Direct sequencing of the amplified enriched regions was performed on the Illumina NextSeq instrument, and the Emory Healthcare GenomOncology Pathology Workbench was used to analyze and annotate variants.
NGS analysis revealed GATA2 c.121C>G (p.P41A) in approximately 49 percent of alleles (Fig. 1F) and FLT3-ITD, 63-bp, with an allele frequency of 22 percent, resulting in 21 amino acid duplication (data not shown). One month after the initial diagnosis, a follow-up marrow sample demonstrated no definite morphologic evidence of APL. Flow cytometric immunophenotyping failed to identify any abnormal hematolymphoid cell populations. Routine chromosome analysis revealed a normal male karyotype. The patient was then initiated on consolidation therapy with ATRA and ATO and completed the therapeutic regimen in March 2022.
Bone marrow biopsy samples from April and July 2022 were hypocellular for age without evidence of residual disease. NGS performed on the April 2022 remission bone marrow demonstrated the disappearance of FLT3-ITD mutation, while the GATA2 c.121C>G (p.P41A) mutation is persistent at an allele frequency similar to that at diagnosis, suggestive of germline origin.
Shortly after achieving remission, the patient developed mild leukopenia secondary to ATRA treatment. This mild leukopenia in combination with his likely germline GATA2 variant led him to be referred for bone marrow transplant and genetic counseling. A pedigree was obtained during the genetic counseling session (Fig. 1G). Very little information is available about the patient’s biological family because he is adopted. He is familiar with his biological mother and unaware of any familial hematological disorders. It was concluded that no additional genetic testing was required for family members, and this variant has minimal impact in making treatment decisions. No plans for bone marrow transplant had been made as of the most recent follow-up in January 2023, and his mild leukopenia is monitored.
Discussion. While this patient’s clinical presentation (leukocytosis and DIC) may appear more consistent with hypogranular APL, these manifestations may also be seen in other AMLs. In addition, the morphology and immunophenotypic profile of the leukemic blasts are not typical of hypogranular APL. Specifically, the leukemic blasts do not display the characteristic bilobed or butterfly-shaped nuclei morphologically. In addition, Auer rods were not identified. Immunophenotypically, the leukemic blasts exhibited low side scatter with expression of many myeloid lineage markers and coexpression of CD34, CD117, and HLA-DR, which is unusual for APL. Although aberrant CD2 expression has been associated with FLT3-ITD in APL,2 it is not specific and is frequently seen in other AMLs.
Patteet, et al., reported a case of hypogranular APL with partial coexpression of CD2, CD34, and HLA-DR.6 Foley, et al., also reported that the presence of CD34 in APL was highly associated with leukocytosis, hypogranular morphology, and coexpression of CD2.7 In addition, a high presenting WBC count (≥ 10 × 109/L) was found to be associated with poor overall survival and increased rate of relapse. However, ultimately, coexpression of CD34 and HLA-DR is extremely rare in APL. The mechanism behind the atypical expression of the lymphoid markers CD2 and HLA-DR remains an area of further investigation.
At diagnosis, next-generation sequencing revealed a genetic variant of strong clinical significance: FLT3-ITD, 63-bp, detected in approximately 22 percent of alleles. FMS-like tyrosine kinase 3 (FLT3) is a proto-oncogene involved in hematopoiesis and is mainly expressed in hematopoietic organs such as bone marrow, thymus, and lymph nodes.8 The FLT3 gene is found in the chromosomal region 13q12.2 and encodes a membrane-bound receptor tyrosine kinase (RTK). The most common form of FLT3 mutation is an internal tandem duplication that promotes ligand-independent auto-phosphorylation and constitutive receptor tyrosine kinase activation, which has been strongly associated with leukocytosis and poor prognosis. In addition, FLT3-ITD is associated with microgranular morphology and involvement of the bcr3 breakpoint of PML.1,8
A genetic variant, GATA2 c.121C>G (p.P41A), was also detected by NGS with allele frequencies of approximately 49 percent and 51 percent in the initial diagnostic and follow-up remission bone marrow samples, respectively. This specific missense variant involves substitution of the nucleotide G for C at position 121 of the GATA2 coding sequence. This sequence variation is predicted to result in change of amino acid at position 41 from proline to alanine near the N terminus before the zinc finger domain, which may alter GATA2 function. While additional testing to confirm the germline nature of the variant (e.g. skin biopsy) was not performed, it is highly probable to be germline in origin given its presence in about 50 percent of tested bone marrow cells before and after remission. Germline mutations in GATA2 are typically found in the zinc finger domains of the GATA2 protein and are associated with increased risk of developing myeloid neoplasms including AML.9 GATA2 is a tumor suppressor gene, and its inactivation by mutation and/or epigenetic silencing has been found to accelerate disease progression in APL and other forms of AML.10


Fig. 1. Morphology and ancillary study results. (A) Blasts in peripheral blood, Wright-Giemsa stain (100×). (B) Blasts on bone marrow biopsy, hematoxylin-eosin stain (40×). (C) Flow cytometry demonstrates coexpression of CD34, CD117, and HLA-DR (86 percent of analyzed cells). (D) Routine chromosome analysis demonstrates abnormal metaphases with a translocation between the long arms of chromosomes 15 and 17, t(15;17)(q24;q21). (E) Interphase FISH results are positive for t(15;17)(PML::RARA). (F) NGS reveals GATA2 c.121C>G (p.P41A) (49 percent variant allele frequency) at initial presentation. (G) Family pedigree with available information.
The GATA2 c.121C>G (p.P41A) variant was first reported in a study by Holme, et al., which investigated genetic heterogeneity in familial myelodysplasia (MDS)/AML.11 The novel variant was identified in a 48-year-old female with MDS in a family with two other individuals also diagnosed with MDS. It has also been reported in an independent study of inherited cytopenias by Kager, et al., who described the variant in a 17-year-old female with moderate neutropenia and B-cell deficiency.12 She experienced recurrent bouts of fever and oral aphthous lesions and was ultimately diagnosed with GATA2 deficiency. Several members of her family were diagnosed with cancer including MDS. The authors concluded that her symptoms were most likely attributed to her heterozygous variant (p.P41A) of GATA2.

While both studies suggest a relationship between this variant and the development of hematological disorders, neither performed functional studies to further characterize this potential association. In addition, a minor allele fraction of the GATA2 c.121C>G (p.P41A) mutation is observed in general population studies with reference to the Genome Aggregation Database (gnomAD).13 In gnomAD, this variant has a population frequency of heterozygotes of approximately 0.06 percent (171 of 276,088 total alleles) across individuals of all age ranges including several known to not have cancer or disease. In the ClinVar database, six labs classified this specific mutation as a variant of uncertain significance, and two labs classified it as likely benign.14 Therefore, further studies are necessary to elucidate the potential significance of this variant in predisposing to the development of hematological disorders.
Conclusion. In summary, this case exemplifies the importance of using a multimodal diagnostic approach to arrive at the final diagnosis. The results of chromosome, FISH, and NGS studies were instrumental in establishing a diagnosis as well as in identifying a germline GATA2 variant [c.121C>G (p.P41A)] in a patient with an unusual APL. The identification of this germline variant also had important implications for known family members as further testing may be necessary. To our knowledge, this is the first description of the presence of this germline variant in a case of an atypical hypogranular APL with coexpression of CD2, CD34, and HLA-DR. This report serves as a contribution to the limited available literature regarding the implications of this unique genetic variant, which remains an area of further investigation.
- Swerdlow SH, Campo E, Harris NL, et al. WHO Classification of Tumours of Haematopoietic and Lymphoid Tissues. 4th rev ed. IARC Press; 2017:30–59.
- Takenokuchi M, Kawano S, Nakamachi Y, et al. FLT3/ITD associated with an immature immunophenotype in PML-RARα leukemia. Hematol Rep. 2012;4(4):e22.
- McReynolds LJ, Calvo KR, Holland SM. Germline GATA2 mutation and bone marrow failure. Hematol Oncol Clin North Am. 2018;32(4):713–728.
- McReynolds LJ, Yang Y, Wong HY, et al. MDS-associated mutations in germline GATA2 mutated patients with hematologic manifestations. Leuk Res. 2019;76:70–75.
- Arber DA, Orazi A, Hasserjian RP, et al. International Consensus Classification of myeloid neoplasms and acute leukemias: integrating morphologic, clinical, and genomic data. Blood. 2022;140(11):1200–1228.
- Patteet L, Vermeulen K, Pieters, et al. A hypogranular variant of acute promyelocytic leukaemia showing a heterogenic immunophenotype with CD34, CD2, HLA-DR positivity: a case report and review of the literature. Acta Clin Belg. 2012;67(1):34–38.
- Foley R, Soamboonsrup P, Carter RF, et al. CD34-positive acute promyelocytic leukemia is associated with leukocytosis, microgranular/hypogranular morphology, expression of CD2 and bcr3 isoform. Am J Hematol. 2001;67(1):34–41.
- Lagunas-Rangel F, Chávez-Valencia V. FLT3-ITD and its current role in acute myeloid leukaemia. Med Oncol. 2017;34(6):114.
- Klco JM, Mullighan CG. Advances in germline predisposition to acute leukaemias and myeloid neoplasms. Nat Rev Cancer. 2021;21(2):122–137.
- Katerndahl CDS, Rogers ORS, Day RB, et al. Tumor suppressor function of Gata2 in acute promyelocytic leukemia. Blood. 2021;138(13):1148–1161.
- Holme H, Hossain U, Kirwan M, Walne A, Vulliamy T, Dokal I. Marked genetic heterogeneity in familial myelodysplasia/acute myeloid leukaemia. Br J Haematol. 2012;158(2):242–248.
- Kager L, Jimenez Heredia R, Hirschmugl T, et al. Targeted mutation screening of 292 candidate genes in 38 children with inborn haematological cytopenias efficiently identifies novel disease-causing mutations. Br J Haematol. 2018;182(2):251–258.
- Genome Aggregation Database; v2.1.1. Accessed Feb. 1, 2023. https://gnomad.broadinstitute.org/gene/ENSG00000179348?dataset=gnomad_r2_1
- National Center for Biotechnology Information; VCV000241716.23. Accessed March 15, 2023. www.ncbi.nlm.nih.gov/clinvar/variation/VCV000241716.23
Dr. An, Dr. Saxe, and Dr. Li are in the Department of Pathology and Laboratory Medicine, and Dr. Vengoechea is in the Department of Human Genetics—all at Emory University School of Medicine, Atlanta.