CAP TODAY and the Association for Molecular Pathology have teamed up to bring molecular case reports to CAP TODAY readers. AMP members write the reports using clinical cases from their own practices that show molecular testing’s important role in diagnosis, prognosis, and treatment. The following report comes from the University of Utah School of Medicine and ARUP Laboratories. If you would like to submit a case report, please send an email to the AMP at amp@amp.org. For more information about the AMP and all previously published case reports, visit www.amp.org.
Karen A. Moser, MD
Tracy I. George, MD
Kristin H. Karner, MD
July 2020—A 77-year-old woman presented at our institution for care with a remote history of lymphoma. Three years earlier she had a bone marrow biopsy performed that by report showed five percent monoclonal kappa restricted plasma cells. At the initial presentation at our institution, serum protein electrophoresis and immunofixation showed an IgA kappa paraprotein (0.52 g/dL), and bone marrow biopsy showed less than five percent plasma cells by CD138 immunohistochemistry that were kappa light chain restricted by flow cytometry, with normal renal function and calcium level. A PET-CT indicated no lytic bone lesions. These findings confirmed the diagnosis of IgA kappa monoclonal gammopathy of undetermined significance (MGUS). Her past medical history was remarkable for chronic oral ulcers for five years of possible autoimmune etiology that were responsive to methylprednisolone, a stroke, hypertension, and hyperlipidemia.
A bone marrow biopsy was performed in September 2018 given several interval changes, including a saddle pulmonary embolus in April 2018, new anemia (hemoglobin 9.9 g/dL, MCV 86.3 fL), and increased serum creatinine (2.08 mmol/L). Flow cytometry, routine cytogenetic analysis, myeloma FISH panel, and myeloid next-generation sequencing panel were requested on this bone marrow biopsy.
Initial bone marrow morphologic findings. In September 2018, CBC and pertinent differential data demonstrated WBC 7.32 × 109/L, RBC 3.75 × 1012/L, hemoglobin 10.0 g/dL, MCV 91.5 fL, platelets 310 × 109/L, and ANC 5.7 × 109/L. The peripheral blood smear was unremarkable with no rouleaux, no dysplasia, and no circulating blasts, mast cells, or plasma cells identified.
The bone marrow aspirate had 0.4 percent blasts, 3.2 percent plasma cells, and a myeloid:erythroid ratio of 4.1. The aspirate smears showed adequate cellular spicules, with normal erythroid and myeloid maturation without significant dyspoiesis, normal megakaryocyte morphology, and singly arranged mature plasma cells. Mast cells were inconspicuous on the bone marrow aspirate smears (Fig. 1).
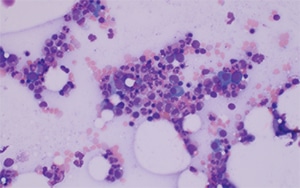
Fig. 1. Bone marrow aspirate smear, 20×. Increased mature plasma cells are evident, but mast cells are not readily identified on the aspirate smear.
The core biopsy and clot section reflected the aspirate smear findings, with 50 percent cellularity, trilineage hematopoiesis, no large sheets or clusters of plasma cells, no areas of fibrosis or spindled cells, and no osteosclerosis on the biopsy. CD138 immunohistochemistry demonstrated less than five percent plasma cells, arranged singly. Flow cytometry demonstrated a small population of monoclonal kappa plasma cells (0.6 percent of leukocytes) that expressed bright CD38, dim to negative CD45, partial CD138, weak CD200, but not CD19, CD20, CD28, CD56, or CD117, similar to previous plasma cell populations described in this patient. No additional abnormal cell populations were identified by flow cytometry.
Cytogenetic and molecular results. Conventional cytogenetic analysis demonstrated a normal female chromosome pattern (46,XX[20]). Myeloma FISH panel reported three copies of CKS1B (1q21) in 50 percent of cells, three copies of CCND1 (11q13) in 7.5 percent of cells, loss of 14q32 involving IGH locus in 81.5 percent of cells, and three copies of PML (15q24) in 4.5 percent of cells scored. Although both the CCND1 and PML alterations were low level, they exceeded the laboratory cutoff value for determining a positive result. The gain of CKS1B and loss of 14q32 were also reported in previous bone marrow specimens for this patient evaluated in our laboratory, but the low-level CCND1 and PML alterations represented new findings.
A myeloid NGS panel was ordered. Briefly, genomic DNA was extracted from bone marrow or peripheral blood leukocytes by the Puregene method (Qiagen), followed by library preparation and enrichment for regions of interest by solution capture (SureSelect, Agilent). Massively parallel sequencing was performed on Illumina MiSeq, HiSeq 2000, or NextSeq platforms. A full list of targeted regions within these genes can be found at http://ltd.aruplab.com/Tests/Pdf/322. Genetic variants were identified by FreeBayes, LoFreq, and Scalpel (single nucleotide variants and small insertions/deletions), and Pindel and Manta (larger insertions/deletions) algorithms. Variants detected were subjected to a rigorous manual curation process including querying variant databases (e.g. SNP database [dbSNP]), the National Heart, Lung, and Blood Institute Exome Sequencing Project, the Exome Aggregation Consortium (ExAC), Catalogue of Somatic Mutations in Cancer (COSMIC), The Cancer Genome Atlas (TCGA), Human Gene Mutation Database (HGMD), and literature review. Oncogenic mutations were defined as either previously reported and/or recurrent hotspot variants, or classical types of variants in certain genes, or novel nonsense, frameshift, canonical ±1 or 2 splice sites and initiation codon variants in genes implicated in myeloid malignancies through acquisition of loss-of-function mutations. The limit of detection for purposes of variant calling was established at one percent VAF, which is defined as the percentage of sequencing reads at a particular location that include the DNA variant in question.
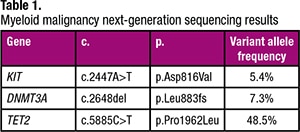 The myeloid NGS panel performed on this bone marrow specimen detected three variants that have been described in myeloid malignancies (Table 1). Specifically, the KIT mutation prompted further morphologic and immunohistochemical evaluation of the bone marrow biopsy, given that KIT mutations are not typically described in plasma cell neoplasms (despite occasional plasma cell neoplasms that aberrantly express CD117) but are observed in the majority of patients with systemic mastocytosis (SM) (particularly the KIT D816V mutation). In fact, activating KIT mutations represent a minor criterion for the diagnosis of SM in the current WHO classification.1
The myeloid NGS panel performed on this bone marrow specimen detected three variants that have been described in myeloid malignancies (Table 1). Specifically, the KIT mutation prompted further morphologic and immunohistochemical evaluation of the bone marrow biopsy, given that KIT mutations are not typically described in plasma cell neoplasms (despite occasional plasma cell neoplasms that aberrantly express CD117) but are observed in the majority of patients with systemic mastocytosis (SM) (particularly the KIT D816V mutation). In fact, activating KIT mutations represent a minor criterion for the diagnosis of SM in the current WHO classification.1
Additional bone marrow morphologic findings. Additional immunohistochemical stains were subsequently performed on the bone marrow biopsy, prompted by the NGS results. Immunohistochemical stain for tryptase showed a mild increase in mast cells (approximately three percent of the total bone marrow cellularity), of which approximately 75 percent were atypical spindled forms with rare dense aggregates of greater than 15 cells. CD25 was positive in about 90 percent of the mast cells (Fig. 2). Repeat evaluation of the H+E-stained bone marrow biopsy showed that the mast cell population was morphologically occult, as we were unable to identify the mast cell population without the aid of immunohistochemical staining. The additional immunohistochemical findings coupled with the NGS data and clinical presentation permitted a diagnosis of indolent SM, in addition to MGUS.
Clinical course. Further clinical evaluation indicated that this patient has no history of skin lesions suggesting cutaneous involvement by mastocytosis, nor did she have hives, wheezing, diarrhea, syncope, or splenomegaly. There was no evidence of B or C findings. Serum tryptase in November 2018 was 63.6 µg/L. She does not require treatment for SM at present, but she will be followed in the hematology clinic at six-month intervals for disease reassessment.
Discussion. SM may be associated with a variety of other hematopoietic neoplasms; in fact, this constitutes a specific category of mast cell neoplasm in the current WHO classification (SM with an associated hematologic neoplasm, SM-AHN). However, most cases of SM-AHN involve associated myeloid neoplasms, most commonly chronic myelomonocytic leukemia.1 SM associated with lymphoid neoplasms is uncommon, and there are few cases reported in the literature of mastocytosis associated with plasma cell neoplasms.2-14 Of these cases, only one is reported to have cutaneous mastocytosis associated with plasma cell myeloma2; the remaining cases are SM associated with plasma cell neoplasms. One of the reported SM patients also had MGUS,6 while two patients had concomitant indolent SM (ISM) and smoldering myeloma9,13; all other reported patients had SM and plasma cell myeloma.
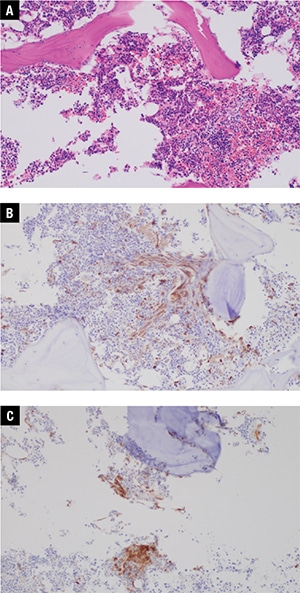
Fig. 2. Bone marrow core biopsy, 20×. A. Mast cells are difficult to identify on H+E stain. B. Immunohistochemistry for tryptase shows spindled mast cells with focal dense aggregates of >15 cells. C. Immunohistochemistry for CD25 shows that the mast cells are positive for CD25.
Another interesting feature of our case, in addition to the unusual association of ISM and MGUS, is the presence of non-KIT mutations. Although KIT mutations are well described in the pathogenesis of mast cell neoplasms and represent a minor criterion for SM diagnosis, information regarding the importance of additional mutations detected in mast cell disease is emerging. Recent retrospective studies evaluating non-KIT mutations by NGS in patients with SM have demonstrated mutations that occur in SM with increased frequency and that may impact disease phenotype or prognosis. Schwaab and colleagues reported additional mutations in 3/12 (25 percent) of ISM patients, with CBL, SETBP1, U2AF1, JAK2, ASXL1, RUNX1, and ETV6 mutations reported in this group.15 This group also noted that in patients with advanced SM but no additional mutations, disease phenotype was less severe, overall survival was improved, and there were not significant cytopenias as compared with patients with one or more non-KIT mutations.15 Pardanani and colleagues evaluated 150 patients with SM, 29 percent of whom had ISM and 53 percent of whom had SM-AHN (nine percent of SM-AHN patients had either lymphoma or myeloma, not further specified).16 In this group, the most commonly mutated genes across all categories of SM were TET2, ASXL1, CBL, SF3B1, DNMT3A, JAK2, U2AF1, and RUNX1, with ASXL1 mutations associated with inferior overall survival and presence of ASXL1 mutations incorporated into a proposed prognostic scoring system for SM patients.16 Of relevance to our case, Pardanani, et al., identified non-KIT mutations (DNMT3A and TET2 only) in 14 percent of ISM patients, essentially identical to our reported case.16 The relationship of the DNMT3A mutation to SM is unclear, as this mutation could represent clonal hematopoiesis of indeterminate potential (CHIP), particularly in elderly individuals. In our case, the TET2 variant with near 50 percent variant allele fraction is likely to be a germline polymorphism. Naumann and colleagues reported another recent study of 109 SM patients (24 percent with ISM), of whom 24 percent had TET2 mutations and one percent had DNMT3A mutations.17 Previous studies looking only at the presence of TET2 and RAS family mutations in SM have suggested some impact on disease phenotype but no clear impact on disease prognosis from these mutations.18,19 Further work is needed to fully characterize the mutational landscape of SM and the prognostic impact, if any, from non-KIT mutations in SM.
In our case, unexpected NGS findings led to an additional diagnosis that would have been missed by routine bone marrow evaluation alone. Our case illustrates the necessity of additional morphologic evaluation to explain and correlate unexpected molecular findings. The identification of unexpected ISM in this case did not prompt immediate treatment, but it contributes to the need for close clinical monitoring for disease progression in this case and will be important information should the patient develop mastocytosis-related symptoms in the future.
- Swerdlow SH, Campo E, Harris NL, et al., eds. WHO Classification of Tumours of Haematopoietic and Lymphoid Tissues. Rev. 4th ed. Lyon, France: IARC Press; 2017.
- Ogg GS, Rosbotham JL, MacDonald DM. Urticaria pigmentosa coexisting with multiple myeloma. Clin Exp Dermatol. 1996;21(5):365–366.
- Hagen W, Schwarzmeier J, Walchshofer S, et al. A case of bone marrow mastocytosis associated with multiple myeloma. Ann Hematol. 1998;76(3–4):167–174.
- Stellmacher F, Sotlar K, Balleisen L, Valent P, Horny HP. Bone marrow mastocytosis associated with IgM kappa plasma cell myeloma. Leuk Lymphoma. 2004;45(4):801–805.
- Sotlar K, Saeger W, Stellmacher F, et al. “Occult” mastocytosis with activating c-kit point mutation evolving into systemic mastocytosis associated with plasma cell myeloma and secondary amyloidosis. J Clin Pathol. 2006;59(8):875–878.
- Pullarkat ST, Sedarat F, Paquette R, Said J. Systemic mastocytosis with plasma cell dyscrasia: report of a case. Leuk Res. 2008;32(7):1160–1163.
- Kluin-Nelemans HC, Ferenc V, van Doormaal JJ, et al. Lenalidomide therapy in systemic mastocytosis. Leuk Res. 2009;33(3):e19–22.
- Motwani P, Kocoglu M, Lorsbach RB. Systemic mastocytosis in association with plasma cell dyscrasias: report of a case and review of the literature. Leuk Res. 2009;33(6):856–859.
- Filanovsky K, Lev S, Haran M, et al. Systemic mastocytosis associated with smoldering multiple myeloma: an unexpected diagnosis in a patient with a rash. Leuk Lymphoma. 2010;51(6):1152–1154.
- Du S, Rashidi HH, Le DT, Kipps TJ, Broome HE, Wang HY. Systemic mastocytosis in association with chronic lymphocytic leukemia and plasma cell myeloma. Int J Clin Exp Pathol. 2010;3(4):448–457.
- Jain P, Verstovsek S, Orlowski RZ, Yap E, Amin HM. An unusual case of aggressive systemic mastocytosis with associated refractory plasma cell myeloma. Clin Lymphoma Myeloma Leuk. 2012;12(6):459–462.
- Butterfield JH, Weiler CR. Systemic mastocytosis in the elderly. Am J Hematol. 2013;88(5):406–408.
- Ghanem S, Garcia G, Ying L, Hurford M, Odaimi M. Systemic mastocytosis with smoldering multiple myeloma: report of a case. Case Rep Oncol Med. 2016;2016:3161768.
- Prabahran AA, Juneja SK. Systemic mastocytosis with concurrent multiple myeloma. Blood. 2018;131(13):1494.
- Schwaab J, Schnittger S, Sotlar K, et al. Comprehensive mutational profiling in advanced systemic mastocytosis. Blood. 2013;122(14):2460–2466.
- Pardanani A, Lasho T, Elala Y, et al. Next-generation sequencing in systemic mastocytosis: derivation of a mutation-augmented clinical prognostic model for survival. Am J Hematol. 2016;91(9):888–893.
- Naumann N, Jawhar M, Schwaab J, et al. Incidence and prognostic impact of cytogenetic aberrations in patients with systemic mastocytosis. Genes Chromosomes Cancer. 2018;57(5):252–259.
- Tefferi A, Levine RL, Lim KH, et al. Frequent TET2 mutations in systemic mastocytosis: clinical, KITD816V and FIP1L1-PDGFRA correlates. Leukemia. 2009;23(5):900–904.
- Wilson TM, Maric I, Simakova O, et al. Clonal analysis of NRAS activating mutations in KIT-D816V systemic mastocytosis. Haematologica. 2011;96(3):459–463.
Dr. Moser is assistant professor of pathology, Dr. George is professor of pathology, and Dr. Karner is assistant professor of pathology, University of Utah School of Medicine and ARUP Laboratories, Salt Lake City.
Test yourself
- Here are three questions taken from the case report.
- Answers
1. What is the most common hematologic neoplasm associated with mastocytosis in the entity systemic mastocytosis with associated hematologic neoplasm (SM-AHN)?
a. Acute myeloid leukemia
b. Chronic myelomonocytic leukemia
c. Follicular lymphoma
d. Plasma cell myeloma
2. The presence of an activating mutation in which gene fulfills a minor criterion for the diagnosis of systemic mastocytosis?
a. ASXL1
b. DNMT3A
c. KIT
d. TET2
3. Mutation in which gene is associated with decreased overall survival in patients with systemic mastocytosis?
a. ASXL1
b. DNMT3A
c. KIT
d. TET2
Answers are online now at www.amp.org/casereports and will be published next month in CAP TODAY.