
CAP TODAY and the Association for Molecular Pathology have teamed up to bring molecular case reports to CAP TODAY readers. AMP members write the reports using clinical cases from their own practices that show molecular testing’s important role in diagnosis, prognosis, and treatment. The following report comes from the University of Washington School of Medicine. If you would like to submit a case report, please send an email to the AMP at amp@amp.org. For more information about the AMP and all previously published case reports, visit www.amp.org.
Hani El Achkar, MD; Marie Das, MD
David Wu, MD, PhD
Daniel Sabath, MD, PhD
October 2025—Chronic lymphocytic leukemia/small lymphocytic lymphoma (CLL/SLL) is a mature B-cell neoplasm composed of small atypical lymphoid cells that often coexpress CD5 and CD23 and are characterized by scant cytoplasm, clumped nuclear chromatin, and indistinct nucleoli. CLL/SLL can involve the peripheral blood, bone marrow, and various lymphoid tissues such as the lymph nodes, tonsils, and spleen, and it may occasionally present in extranodal locations as well.1 Within involved lymph nodes, pale-staining proliferation centers consisting of prolymphocytes or paraimmunoblasts are a characteristic finding in CLL/SLL.
CLL/SLL makes up 25 to 30 percent of total leukemias in the United States, predominantly affecting older adults with an average age of 70 and demonstrating a slightly higher incidence in male patients.2 The clinical course of CLL/SLL is heterogeneous, ranging from indolent to aggressive disease. Cytogenetic and molecular features play a crucial role in the initial prognostication of CLL/SLL, and it is important to consider that certain molecular markers can evolve over the course of the disease, potentially leading to drug resistance and disease progression. Covalent Bruton tyrosine kinase (BTK) inhibitors, such as ibrutinib, have become a cornerstone of CLL/SLL therapy, used both as first-line treatment and for managing disease progression. However, mutations in the BTK gene can result in acquired resistance to these therapies, making it essential to assess a patient’s BTK mutation status for effective treatment planning.
Here, we present two male patients with CLL/SLL who experienced disease progression and were found to have a similar mutational profile, with each patient having the rare occurrence of two independently arising resistance clones, both of which resulted in the BTK p.C481S ibrutinib resistance mutation.
Case No. 1. A 75-year-old male smoker presented in 2019 with bilateral cervical lymphadenopathy. CBC showed leukocytosis with absolute lymphocytosis. CT revealed extensive lymphadenopathy above and below the diaphragm, including a 7.4-cm retroperitoneal nodal conglomerate and mild splenomegaly. An excisional lymph node biopsy confirmed CD5-positive B-cell non-Hodgkin lymphoma, consistent with CLL/SLL. Flow cytometry showed an abnormal B-cell population with bright CD5 and CD200, intermediate CD22, five percent CD38, dim kappa restriction, decreased CD20, and negative CD3, CD10, and CD56. Subsequent bone marrow biopsy confirmed CLL with del(17p) and trisomy 12 on FISH and a complex karyotype. Ibrutinib therapy induced remission but caused atrial fibrillation, prompting a switch to a well-tolerated acalabrutinib in 2021.
The patient was recently hospitalized for increased fatigue and abdominal pain, with laboratory studies revealing elevated bilirubin levels (total: 5.2 mg/dL, direct: 2.7 mg/dL), AST (184 U/L), and ALT (282 U/L), while abdominal MRI showed new lymphadenopathy (up to 2 cm) and hepatomegaly. Peripheral blood flow cytometry identified an abnormal B-cell population (33.8 percent of leukocytes) expressing CD5, consistent with persistent disease. Subsequent lymphoid gene panel testing revealed clinically significant variants, including NOTCH1 (p.P2514Rfs*4, NM_017617.3:c.7541_7542del, VAF 16 percent), two distinct BTK variants (p.C481S, NM_000061.3:c.1441T>A, VAF 11 percent, and p.C481S, NM_000061.3:c.1442G>C, VAF 10 percent), and TP53 (p.R110P, NM_000546.5:c.329G>C, VAF three percent). The C481S mutations, associated with resistance to covalent BTK inhibitors like ibrutinib and acalabrutinib, necessitated a change in therapy to obinutuzumab and venetoclax.
Case No. 2. A 65-year-old male patient known to have chronic lymphocytic leukemia characterized by unmutated IGHV and a complex karyotype was diagnosed in 2014 and managed initially with a watch-and-wait strategy for four years. In 2018, due to rising WBC counts and symptomatic lymphadenopathy, the patient was enrolled in protocol 9905 and randomized to full-dose zanubrutinib (160 mg BID). Treatment was well tolerated with minimal toxicity, and CBC showed no lymphocytosis (ALC <4000) or cytopenias, indicating an excellent response.
Six years later, laboratory tests showed declining platelets (141k), neutropenia (ANC 1.36), and rising ALC (8.46). The patient remained asymptomatic but had palpable lymphadenopathy, including an approximately 1-cm right cervical and an approximately 2-cm left axillary node, suggesting disease progression. Molecular testing for BTK single-gene analysis revealed two variants: p.C481S, NM_000061.3:c.1441T>A, VAF 36 percent, and p.C481S, NM_000061.3:c.1442G>C, VAF 12 percent. No clinically significant variants were identified in PLCG2. With evidence of progression and based on molecular results, zanubrutinib was stopped, and the patient started on obinutuzumab and venetoclax.
Both cases highlight the evolution of CLL/SLL, with BTK C481S mutations driving resistance and necessitating alternative therapeutic strategies, such as obinutuzumab-venetoclax–based regimens, to manage relapsed/refractory CLL. Fortunately, both patients are currently doing well clinically and remain on obinutuzumab and venetoclax therapy. For the patient in case No. 1, the current line of therapy has resulted in resolution of his lymphadenopathy and a normal white blood cell count. In the event of disease progression, next-line therapy options would include pirtobrutinib, CAR T-cell therapy, or clinical trials. The patient in case No. 2 was able to decrease the frequency of his surveillance visits from monthly to every three months given his stable clinical status.
Methods. Both samples were evaluated using a multigene next-generation sequencing panel designed to detect mutations associated with hematologic disorders. Purified genomic DNA was enzymatically fragmented, and targeted sequences were isolated by hybrid capture probes and sequenced on an Illumina NGS platform. The nucleic acid extraction, amplification, sequencing, and data interpretation are performed in the UW molecular hematopathology laboratory. A custom bioinformatics pipeline developed by the UW NGS analytics laboratory was used for data analysis as previously described.3
The samples from these two patients were processed on different days and were on different runs of this NGS assay, minimizing concerns about sample contamination.
Discussion. Some CLL/SLL patients experience indolent disease and may not require treatment for many years, while others may face rapid progression of disease. Clinical features, such as the degree of lymphocytosis and the presence of hepatomegaly or splenomegaly, anemia, and thrombocytopenia, as well as molecular and cytogenetic features—including evaluation of del(11q), del(13q), del(17p), trisomy 12, TP53 mutation analysis, and IGHV gene mutation status—play a critical role in staging and prognostication for patients with newly diagnosed CLL/SLL.1
Prior to the advent of BTK inhibitors, the mainstay of treatment for CLL/SLL was typically a combination of chemotherapy and immunotherapy. BTK is a kinase involved in B-cell receptor signaling and is upregulated in CLL/SLL cells compared with normal B cells. Targeted inhibition of BTK blocks B-cell receptor signaling via covalent binding of the BTK C481 residue, thus diminishing proliferation signals in the neoplastic cells.4,5 This blockade has been shown to translate clinically into improved survival outcomes for CLL/SLL patients treated with BTK inhibitors compared with those receiving prior standard therapies.6 The approval of the BTK inhibitor ibrutinib in 2014 for patients who had received at least one prior therapy, and in 2016 for untreated CLL, has dramatically transformed the therapeutic landscape for CLL/SLL.
BTK inhibitor therapy has been shown to lead to durable remission in many CLL/SLL patients. In fact, long follow-up from the pivotal RESONATE-2 study demonstrated that the estimated five-year progression-free survival rate was 70 percent for patients treated with first-line ibrutinib and 12 percent for those treated with chlorambucil.7 Follow-up at eight years demonstrated continued benefit with ibrutinib therapy, with a PFS rate of 59 percent for ibrutinib and nine percent for chlorambucil.8 Notably, the improvement in PFS seen with first-line ibrutinib as compared with chlorambucil was consistent across CLL patient groups, including those with high-risk genetic features, such as del(11q), TP53 mutation, and/or unmutated IGHV, and those with advanced-stage disease.8
Although many patients are able to achieve long-term remission with BTK inhibitor therapy, a subset of patients will still develop progressive disease after an initial response to therapy due to acquired resistance to ibrutinib (or other BTK inhibitors). Resistance to targeted BTK inhibition therapy has been attributed to the development of resistance mutations in certain key genes, most commonly involving BTK and/or PLCG2. Within the BTK gene, the most common resistance mutations are p.C481S (substitution of cysteine with serine) and p.C481R (substitution of cysteine with arginine). These alterations interfere with the ability of ibrutinib to covalently bind to and diminish BTK activity.9 Similarly, zanubrutinib and acalabrutinib also rely on covalent binding to the C481 residue to exert their effects. As a result, the common p.C481S and p.C481R mutations also confer resistance to these second-generation BTK inhibitors.
Pirtobrutinib is a third-generation BTK inhibitor that does not rely on covalent binding of the C481 residue; rather, it noncovalently/reversibly inhibits BTK within the adenosine triphosphate-binding site. Pirtobrutinib was found to be a promising treatment option for patients with pretreated CLL, including those with a BTK C481 resistance mutation.10,11 Patients with progressive disease on pirtobrutinib therapy have been found to have resistance mutations other than the canonical C481 mutations, such as T474 codon mutations and L528W.12 PLCG2 is downstream of BTK in the B-cell receptor signaling pathway, and resistance mutations occurring in PLCG2 tend to be gain-of-function alterations that allow for continued signaling within the B-cell receptor pathway despite BTK blockade.9,15 Different BTK inhibitors exhibit varying levels of specificity against resistance mutations, influencing their efficacy. For example, ibrutinib demonstrates the widest range of coverage against non-C481 mutations, while acalabrutinib is potent against the L528W mutation but less effective against T474I, and zanubrutinib is inactive against L528W but modestly effective against T474I.13 These findings underscore the importance of BTK gene sequencing during a patient’s treatment course, particularly when disease progression is suspected, with the ability to detect not only resistance mutations at the common C481 site but also those at other locations within the gene.14
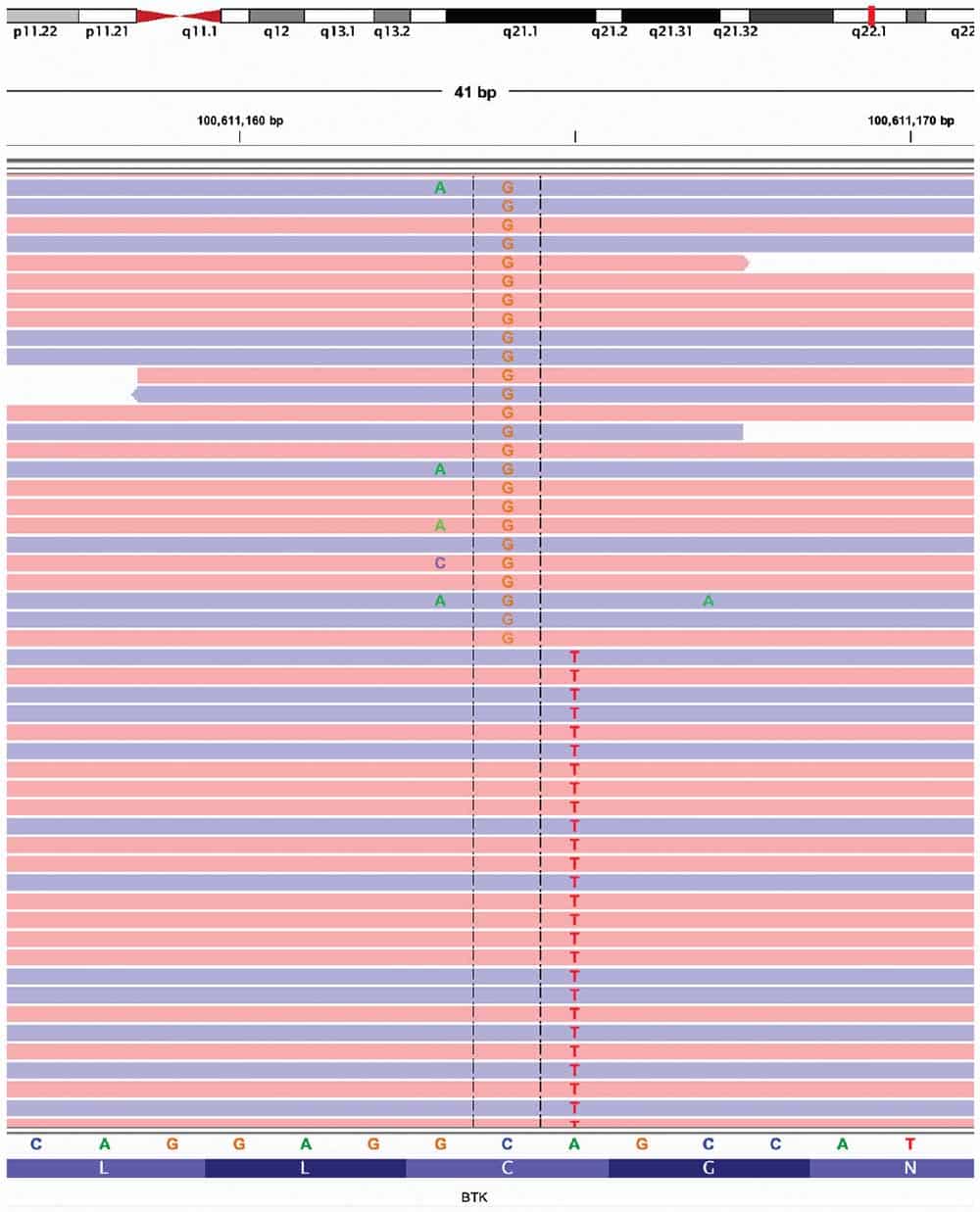
Regarding the two patients described in this report, upon clinical disease progression, both were found to have the common BTK p.C481S mutation, which causes resistance to covalent BTK inhibitors such as ibrutinib, acalabrutinib, and zanubrutinib. Notably, each patient had two separate mutations, both of which resulted in the BTK p.C481S mutation. The first was a T>A substitution at nucleotide 1441 (NM_000061.3:c.1441T>A), and the other a G>C substitution at nucleotide 1442 (NM_000061.3:c.1442G>C). Based on review of the sequencing data in Integrative Genomics Viewer (IGV), the two BTK variants were never located on the same read with one another (“not in phase”), meaning they did not occur in the same physical BTK allele. The BTK gene is located on the X chromosome; therefore, if the two patients had been female with two X chromosomes, observing such a pattern in IGV could mean that the two variants were within the same cell but in trans (on different alleles), or involving two different cell clones. Since both of our patients were male, with only one X chromosome each, the most likely explanation for the variants not being in phase is that they are present within two different independently arising clonal populations.
The presence of two independently arising BTK p.C481S mutations is a pattern of resistance mutations that has not been commonly reported. There have been reports of relapsing CLL/SLL patients harboring more than one BTK mutation; however, the mutations have been different from one another (for example, BTK p.C481S co-occurring with BTK p.T474I, p.C481R, or p.C481Y).16-19 The clinical impact of two separate clonal populations, each with a BTK p.C481S mutation, as observed in the two patients described in this report, compared with the impact of p.C481S in conjunction with a different BTK mutation, is unclear. However, it can be assumed that the presence of any two BTK resistance mutations likely contributes to increased drug resistance.
In summary, the patients described in this report show a unique pattern of BTK resistance mutations, with two different, independently arising clonal populations. Further studies are needed to better understand the clinical significance of this pattern of BTK mutations, and their role in drug resistance, which may help guide more effective treatment strategies for CLL/SLL patients.
- Naresh KN, Ferry JA, Rossi D, et al. Chronic lymphocytic leukaemia/small lymphocytic lymphoma. In: WHO Classification of Tumours Editorial Board, ed. Haematolymphoid Tumours. Vol. 11. IARC Press; 2024:368–377. WHO Classification of Tumours; 5th ed.
- Mukkamalla SKR, Taneja A, Malipeddi D, Master SR. Chronic lymphocytic leukemia. In: StatPearls. StatPearls Publishing. Updated March 7, 2023. https://www.ncbi.nlm.nih.gov/books/NBK470433/
- Kuo AJ, Paulson VA, Hempelmann JA, et al. Validation and implementation of a modular targeted capture assay for the detection of clinically significant molecular oncology alterations. Pract Lab Med. 2020;19:e00153.
- Woyach JA, Ruppert AS, Guinn D, et al. BTKC481S-mediated resistance to ibrutinib in chronic lymphocytic leukemia. J Clin Oncol. 2017;35(13):1437–1443.
- Woyach JA, Bojnik E, Ruppert AS, et al. Bruton’s tyrosine kinase (BTK) function is important to the development and expansion of chronic lymphocytic leukemia (CLL). Blood. 2014;123(8):1207–1213.
- Burger JA, Tedeschi A, Barr PM, et al. Ibrutinib as initial therapy for patients with chronic lymphocytic leukemia. N Engl J Med. 2015;373(25):2425–2437.
- Burger JA, Barr PM, Robak T, et al. Long-term efficacy and safety of first-line ibrutinib treatment for patients with CLL/SLL: 5 years of follow-up from the phase 3 RESONATE-2 study. Leukemia. 2020;34(3):787–798.
- Barr PM, Owen C, Robak T, et al. Up to 8-year follow-up from RESONATE-2: first-line ibrutinib treatment for patients with chronic lymphocytic leukemia. Blood Adv. 2022;6(11):3440–3450.
- Woyach JA, Furman RR, Liu TM, et al. Resistance mechanisms for the Bruton’s tyrosine kinase inhibitor ibrutinib. N Engl J Med. 2014;370(24):2286–2294.
- Thompson PA, Tam CS. Pirtobrutinib: a new hope for patients with BTK inhibitor-refractory lymphoproliferative disorders. Blood. 2023;141(26):3137–3142.
- Mato AR, Shah NN, Jurczak W, et al. Pirtobrutinib in relapsed or refractory B-cell malignancies (BRUIN): a phase 1/2 study. Lancet. 2021;397(10277):892–901.
- Wang E, Mi X, Thompson MC, et al. Mechanisms of resistance to noncovalent Bruton’s tyrosine kinase inhibitors. N Engl J Med. 2022;386(8):735–743.
- Woyach JA, Ghia P, Byrd JC, et al. B-cell receptor pathway mutations are infrequent in patients with chronic lymphocytic leukemia on continuous ibrutinib therapy. Clin Cancer Res. 2023;29(16):3065–3073.
- Tam CS, Balendran S, Blombery P. Novel mechanisms of resistance in CLL: variant BTK mutations in second-generation and noncovalent BTK inhibitors. Blood. 2025;145(10):1005–1009.
- Liu TM, Woyach JA, Zhong Y, et al. Hypermorphic mutation of phospholipase C, γ2 acquired in ibrutinib-resistant CLL confers BTK independency upon B-cell receptor activation. Blood. 2015;126(1):61–68.
- Woyach JA, Huang Y, Rogers K, et al. Resistance to acalabrutinib in CLL is mediated primarily by BTK mutations. Blood. 2019;134(suppl 1):504.
- Sun C, Mali R, Kositsky R, et al. Extended follow-up and resistance mutations in CLL patients treated with acalabrutinib. Blood. 2023;142(suppl 1):1891.
- Brown J, Mashima K, Fernandes S, et al. Mutations detected in real world clinical sequencing during BTK inhibitor treatment in CLL. Res Sq. Preprint posted online Jan. 16, 2024. doi:10.21203/rs.3.rs-3837426/v1
- Woolston DW, Lee ND, Shadman M, et al. Ultra-deep mutational landscape in chronic lymphocytic leukemia uncovers dynamics of resistance to targeted therapies. Haematologica. 2024;109(3):835–845.
Dr. Wu is professor and director of the molecular genetic pathology fellowship program, and Dr. Sabath is professor and head of the Hematology Division, both in the Department of Laboratory Medicine and Pathology, University of Washington School of Medicine. At the time of the writing of the report, Dr. El Achkar and Dr. Das were molecular genetic pathology fellows in the UW Department of Laboratory Medicine and Pathology. Dr. El Achkar is now a hematopathology fellow, Department of Pathology, Molecular and Cell-based Medicine, Icahn School of Medicine at Mount Sinai. Dr. Das is now assistant professor, Department of Pathology and Laboratory Medicine, University of Louisville.