
Kevin B. O’Reilly
October 2014—The Food and Drug Administration’s plan to subject many laboratory-developed tests to a new layer of regulatory requirements over the course of the next decade is drawing sharply contrasting reactions from stakeholders who view it as either an essential step to improve patient safety or a hindrance that will stifle diagnostic innovation and test improvement.
Despite that fundamental disagreement on the policy’s substance, there does seem to be consensus among informed observers that the FDA is determined to take action, that legislative intervention to block the agency faces long odds, and that the agency’s final guidance will create a regulatory challenge for labs unrivaled by anything out of Washington since CLIA ’88.
The FDA framework was unveiled in detail July 31 as part of a statutorily required notification to Congress 60 days prior to publication as a draft guidance. The agency on Oct. 3 gave notice in the Federal Register that the draft guidance was publicly available. The 41-page document—“Draft Guidance for Industry, Food and Drug Administration Staff, and Clinical Laboratories: Framework for Regulatory Oversight of Laboratory Developed Tests (LDTs)”—can be found at http://j.mp/ldtdraft.

The FDA’s plan has sparked concern among some organizations within lab medicine and raised worries about how the agency will answer key questions that could have significant consequences. These include what counts as a laboratory-developed test, which tests will fall under the premarket review requirement, and how requirements for good manufacturing practices—heretofore applied solely to medical device manufacturers and known as quality system regulations—will be applied to clinical labs and harmonized with existing CLIA requirements.
“It’s now clear that the FDA will no longer take a hands-off approach,” said Richard S. Cooper, manager of the National Healthcare Practice Group at the law firm of McDonald Hopkins. Cooper said the FDA’s proposed changes—while not as far-reaching as CLIA because they would only apply to labs with LDTs—could represent the biggest change to laboratory regulation since that landmark federal legislation.
“This is the first step in a change in approach. And it’s a fairly comprehensive, and fairly material, change,” Cooper said in a Sept. 17 webinar hosted by The Dark Report. “Obviously, if the FDA goes forward, it intends to take a very meaningful, very robust role in regulating laboratory-developed tests.”
In the same webinar, Jane Pine Wood predicted that final guidance won’t arrive sooner than the summer of 2015. Wood is a colleague of Cooper’s at McDonald Hopkins and represents clinical, anatomic, molecular, and toxicology laboratories. She advised lab leaders not to delay preparation given the likelihood of some kind of change in LDT oversight.
“This is going to be a whole new ballgame for a lot of labs,” she said. “This is going to push them into an area where they may not have any experience, or may not have the appropriate personnel to assist. The earlier you get started on the process, and the earlier you get educated about what’s proposed and what ultimately comes down the road, the earlier you can take the appropriate steps.”
Laboratory leaders should review which LDTs they are offering and evaluate where they are likely to fit within the FDA’s proposed oversight scheme, Cooper added.
“You might also think about which LDTs you want to consider bringing online before the effective date of the final guidance,” he said. “You’re also going to need personnel or consultants who are versed in FDA requirements so they can step into the role of FDA compliance advisor and help make sure you’re compliant from that standpoint. We may see some of our lab clients investing in new hires that have FDA experience, likely drawn from the medical device field since that’s the category under which these tests are viewed as falling.”
Labs also should assess the likely cost and compliance impact of the FDA’s plan on their operations, Wood said.
“It’s no longer a free-market situation where these facts are strictly determined based on internal capabilities,” she said. “You now have to answer to an oversight agency, and that will have a material impact on your timetables.”
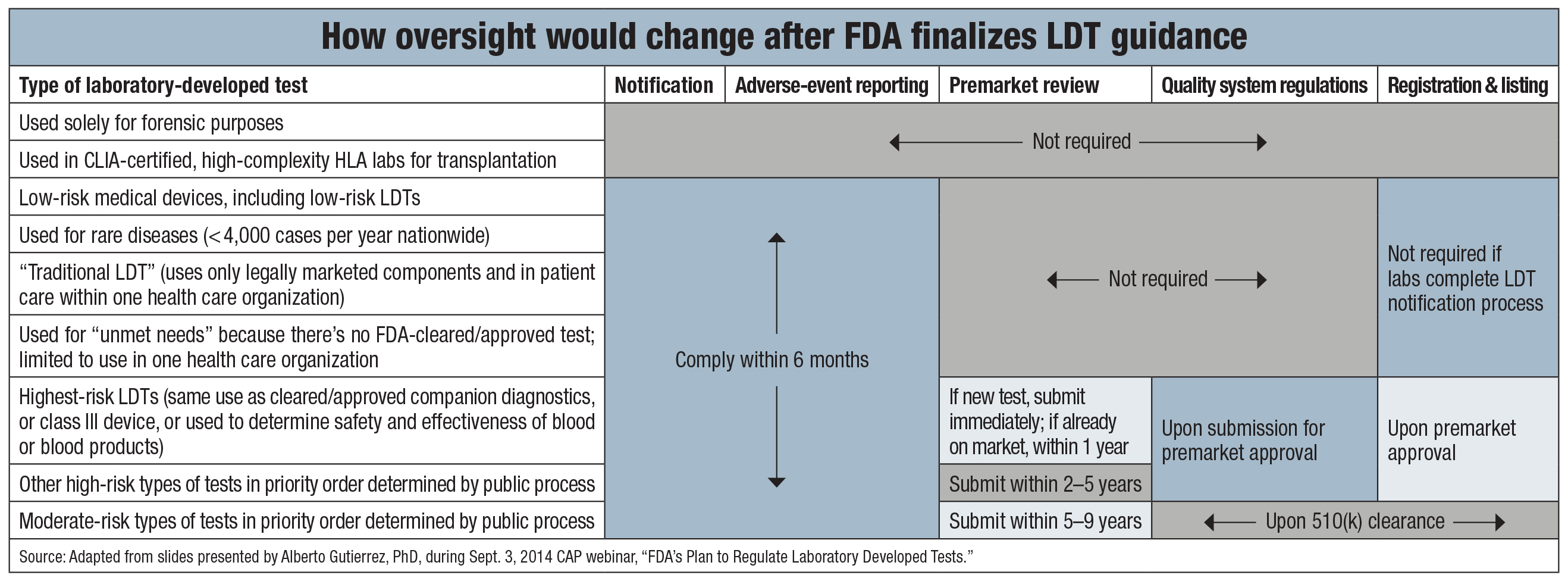
Under the FDA plan, labs would have to tell the agency about most of their LDTs through a formal notification process that is spelled out in a separate, 28-page Oct. 3 draft guidance available at http://j.mp/ldtnotification. Lab-developed tests used in forensics, histocompatibility, stem cell, or tissue transplantation laboratories would be exempt from the notification and other requirements. Other tests deemed to pose low risk would not be required to get premarket review, and neither would tests used for rare diseases.
“Traditional LDTs”—the FDA’s term for tests that use only legally marketed components, require nonautomated interpretation, and are used in the care of patients within a single health care organization—also would fall into the low-risk category. Tests in this category would receive regulatory treatment similar to what is given to class I medical devices.
High-risk LDTs would be treated like class III medical devices, meaning requisite premarket review within one to five years after the FDA guidance is finalized. The agency said it will use expert advisory panels to help classify tests based on risk and various factors (“What makes a high-risk LDT?” page 6).

The FDA will give its highest priority to reviewing LDTs with the same intended use as companion diagnostics or class III medical devices that have already received the agency’s approval or clearance. Also, certain LDTs used to determine the safety and effectiveness of blood or blood products would be subject to the agency’s highest priority review. Tests in the highest-priority category would have to be submitted for premarket review within a year after the FDA finalizes its LDT guidance.
Another category of lab-developed tests, those deemed to be of moderate risk, would have to be submitted for 510(k) clearance within five to nine years of final guidance from the FDA. The agency said it plans to use “accrediting third parties to carry out review of most moderate-risk LDTs requiring a premarket notification.”
The FDA’s tiered, risk-based framework is similar to the oversight approach the CAP outlined in April 2010. The College’s regulatory model (http://j.mp/cap-ldtapproach) seeks “targeted FDA review and approval of clinical claims for only high-risk LDTs, with oversight of compliance by laboratories performing high-risk LDTs by CMS and CMS-deemed accreditors.”
The CAP met with the FDA to clarify sections of the agency’s proposed guidance.
“The CAP will further evaluate the proposed oversight guidance and continue discussions with the FDA and key stakeholders,” CAP president Gene N. Herbek, MD, said in a statement. “The College will also be vigorously engaged during the public comment period and other forums to ensure the final guidance documents improve patient care, meet public health needs, and ensure access to accurate and safe LDTs.”
“The CAP will work to ensure LDT oversight assures quality laboratory testing for patients in a manner that is consistent with principles outlined by the CAP,” Dr. Herbek added. “The proposed FDA guidance embodies a number of those key principles. Where there are differences, the CAP will work with stakeholders so requirements do not impede innovation or increase administrative burden on laboratories. The CAP will provide its recommendations and propose changes to improve the guidance during the public hearing and comment period.”
Alberto Gutierrez, PhD, director of the FDA’s Office of In Vitro Diagnostics and Radiological Health, said in a Sept. 3 CAP webinar that the agency would hold a public meeting “sometime in early January” of 2015. According to the agency’s Federal Register notice, comments on the draft guidance should be submitted by Jan. 31, 2015. That notice also includes instructions on how to comment and is available at http://federalregister.gov/a/2014-23586.
“We don’t really know, or have a lot of control over, how much time the process will take,” Dr. Gutierrez said. “Sometimes it’s highly unpredictable. This draft guidance took four years. I don’t think it will be that long, but I don’t have a good hold on how long that will take.”
The timing of the FDA’s final LDT guidance could be critical for labs whose tests would be subject to the agency’s new enforcement plan, according to Wood.
“This is an important takeaway,” she said in The Dark Report webinar. “If you have an existing LDT and are likely in the higher-risk category where premarket review is required, you definitely want to file the notification in the six months after the final guidance. What that means is your LDT is already on the market. So, until the FDA comes back to you and says, ‘Time’s up; you’ve got to file for premarket review,’ you can go ahead and manufacture your LDT and provide it to the public. But after the guidance is finalized, you can’t put it on the market until you have the premarket review.”
Outside of the FDA’s fairly explicit warning on which LDTs will get the highest priority review, the agency’s risk-classification scheme paints with a broad brush. In its notice to Congress, the FDA said it would be another 18 months after final guidance on LDTs before the agency would issue subsequent draft guidance with further details “to describe what the agency considers generally to be class I, II, or III.”
That opacity is part of what bothers Edward R. Ashwood, MD, about the FDA’s plan to regulate LDTs. He is president and CEO of ARUP Laboratories, which by his count offers “well more than 1,000” LDTs to its customers.
“They say they’re going to take a year or more to figure this out, and establish an advisory committee to give us advice on what category your device falls into. Well, who does the risk analysis?” Dr. Ashwood tells CAP TODAY. “When I see how they’ve classified tests so far, that’s where I become skeptical.”

Dr. Ashwood and 22 other lab medicine leaders from the Mayo Clinic, Brigham and Women’s Hospital, Stanford University Medical Center, and other prestigious academic medical centers wrote a letter this summer asking the White House Office of Management and Budget to put a stop to the FDA’s moving forward with its LDT framework. That did not happen, much to Dr. Ashwood’s dismay.
The FDA’s plan is “as bad as it could be for the future of laboratory medicine and pathology,” he says. “They are converting almost all clinical laboratories into manufacturers.”
He notes that the agency said that significant modifications to already approved devices will be considered LDTs and would receive greater scrutiny as such under its plan.
“What’s a major modification? If you’re using a robot to do the manual steps indicated in a test kit, does that count? What if you’re changing the reagent volumes specified in the kit, or replacing a buffer with a better buffer? Where does it stop?”
“Quite frankly,” Dr. Ashwood says, “we use a lot of FDA-approved kits but without following the standard operating instructions that come with those kits. We modify them and make them better.”
Another burdensome element of the FDA plan, according to Dr. Ashwood, is that when 510(k) submission is required, that would entail stating an LDT’s intended use.
“We don’t do that now,” he says. “Oftentimes, a laboratory-developed test’s intended use changes with time. The more you know, the more intended uses there are. It’s hard to specify that intended labeling up front and never deviate from it. It’s going to cost a lot, and it’s going to be very resource intensive, and not make anything safer or anything more effective.”

Roger D. Klein, MD, JD, chair of the Association for Molecular Pathology’s Professional Relations Committee, also objected strongly to elements of the FDA’s LDT plan. In a position statement, the AMP has said lab-developed tests should be called “laboratory-developed procedures,” arguing that the term better captures their nature as medical services as opposed to medical devices (Ferreira-Gonzalez A, et al. J Mol Diagn. 2014;16[1]:3–6).
“It seems that, were this document to take effect, it’s likely to remove lab-developed procedures from most laboratories whenever there’s an IVD available for the same use. It doesn’t seem like it would make sense, financially, for a lab to offer an LDT,” Dr. Klein said in a Sept. 16 webinar hosted by the AMP. “This draft framework could result in exclusive use of FDA-approved kits. For those who do oncology, what does this mean for next-generation sequencing? Many organizations are doing NGS panels for cancer testing. If one has to use the FDA-cleared or -approved diagnostics, that’s removing from the NGS panels the tests that typically provide some reimbursement for those panels. . . . That could have an enormously destructive impact on the ability to perform next-generation sequencing of panels in oncology.”
Another open question, Dr. Klein said, was whether the FDA’s “unmet needs” exemption that cites intra-health system testing as a factor in determining its applicability would pertain to hospital outreach programs. He estimated that securing premarket approval for some LDTs could cost millions.
“And I don’t think 510(k) will be inexpensive, either,” he said. “The cost of doing so will probably substantially exceed the resources that most clinical labs are willing to expend on a particular test.”
In an email to CAP TODAY, Dr. Klein underscores his view that the FDA’s framework for LDTs would present a thoroughgoing challenge for lab medicine.
“We should be clear that this is not about creating a bunch of new regulations to oversee LDTs—FDA could not do that without notice-and-comment rulemaking. It is about designating laboratory-developed tests as medical devices, and clinical laboratories as medical device manufacturers,” Dr. Klein says. “The change is profound.”
Clinical geneticist and pediatrician Wendy K. Chung, MD, PhD, echoes Dr. Klein’s concerns about the FDA’s plan.

“When the FDA regulates, the whole thing gets locked down—the informatics, the reagents. There may be relatively minor tweaks, but sometimes there are much bigger things in terms of informatics improvement,” Dr. Chung tells CAP TODAY. “We’re just in too dynamic a phase in terms of understanding genomics that it’s just going to paralyze us.”
“If it’s three new actionable genes you find out about each year, that’s three times a year to go through the FDA to get revalidated and reapproved,” adds Dr. Chung, who directs the clinical cancer genetics program at Columbia University Medical Center. “It becomes untenable in terms of being able to keep pace with that. If we were 10 years from now, that’s a different issue than when we are in this very dynamic phase of discovery. We clearly want to transfer this knowledge into patient care as quickly as possible. We want the best care at the lowest cost, but we can’t achieve that with one hand tied behind our back.”
In the CAP webinar, the FDA’s Dr. Gutierrez tried to ease concerns about risk classification and the potential costs and time delay associated with premarket review.
“If you look at the in vitro diagnostics that have been regulated in the last 30-some years, you can see that about half of the IVDs—and I think the CAP risk-based scenarios fit fairly well with the way we look at it—are class I, or low risk,” he said. “About 45 percent of the rest are class II, and about five percent of the IVDs we look at are class III. It’s really a small proportion of them.”
Moreover, “the agency rarely uses prospective clinical trials for most of the things we approve,” he said. “I can think of a couple of premarket approvals where we have done clinical trials, but they are really not that usual. Lots of times we use retrospective data.” Dr. Gutierrez added that for 510(k) submissions, “we do allow people to use literature. The issue with the literature is tying the performance of your device to the one in the literature. Lots of times that can be bridged, and literature data is acceptable.”
The broader question of what regulatory burden labs will face as a result of the FDA’s LDT plan remains a big unknown.
 In its Federal Register notice, the FDA said it expects approximately 650 laboratories will be required to provide notification about 17 LDTs each in the first year after guidance is finalized. In New York alone, 565 labs submitted 9,800 lab-developed tests for review under that state’s law in 2014. The FDA noted that the National Institutes of Health Genetic Test Registry includes about 7,600 tests that are not FDA-approved or -cleared.
In its Federal Register notice, the FDA said it expects approximately 650 laboratories will be required to provide notification about 17 LDTs each in the first year after guidance is finalized. In New York alone, 565 labs submitted 9,800 lab-developed tests for review under that state’s law in 2014. The FDA noted that the National Institutes of Health Genetic Test Registry includes about 7,600 tests that are not FDA-approved or -cleared.
“If we assume that genetic tests represent roughly 70 to 80 percent of all LDTs, this supports our estimate of 11,050 LDTs (total annual responses in the first year),” said the agency’s Federal Register notice.
However, in testimony given at the House Energy and Commerce Committee’s Sept. 9 Health Subcommittee hearing on lab-developed tests, M. Kathleen Behrens Wilsey, PhD, noted that the CMS says more than 11,000 CLIA-certified labs perform high-complexity testing, including LDTs. Dr. Behrens Wilsey is co-founder of the Coalition for 21st Century Medicine, whose members include Foundation Medicine, Myriad, and Prometheus.
“We estimate that many molecular markers are offered as LDTs by hundreds, if not thousands, of laboratories across the country. This would translate into potentially tens of thousands of premarket submissions to the FDA,” Dr. Behrens Wilsey added. She said the FDA’s framework “leaves far too many critical questions unanswered.”
The American Medical Association also submitted testimony to the Health Subcommittee that held the hearing, reiterating its opposition to the FDA framework because it would “severely limit patient access to life-saving tests.”
Another organization that has been steadfast in its opposition to FDA oversight of LDTs is the American Clinical Laboratory Association. ACLA in June 2013 filed a citizen petition challenging the FDA’s statutory authority to regulate LDTs. The trade group says the agency is exceeding the letter of the law, and that LDT oversight should be restricted to the CMS under CLIA.
So, is ACLA ready to take the FDA to court over the matter?
“We haven’t made any decisions or determination about litigation at this point,” ACLA president Alan Mertz tells CAP TODAY. He says that even if the FDA did have the authority to oversee LDTs—and ACLA believes it does not—the agency should use the rulemaking process, rather than guidance, to do so.
“In rulemaking, the agency has to do an economic analysis, and the economic analysis would include the economic impact on the lab industry as well as on the FDA—and what resources it would take the FDA to do this,” he says. “In order to do that, the agency would have to define up front which LDTs would be subject to regulation. You have to lay out which are the high-risk tests, and how widespread this is going to be.”
Mertz also says the FDA has failed to “explain how they would harmonize FDA regulation of laboratories with CLIA regulation that labs already have. There’s a tremendous amount of duplication. . . . The rulemaking would require the FDA to lay out how this would be done.”
While labs may be apprehensive about the impact of the FDA’s plan to oversee LDTs, many stakeholders are welcoming the agency’s action after years of debate, consideration, and delay.
At the Sept. 9 congressional hearing, physicians speaking on behalf of the American Heart Association and the American Association for Cancer Research spoke in support of the FDA plan.
“We commend the FDA for taking a regulatory approach that puts patients first by proposing a classification of LDTs based on risk posed to the patient,” Charles L. Sawyers, MD, said in his testimony. Dr. Sawyers, immediate past president of the AACR, is chair of the human oncology and pathogenesis program at Memorial Sloan Kettering Cancer Center.
“The proposed framework strikes a thoughtful balance between protecting patient safety while promoting research and innovation,” Dr. Sawyers added. He said the nine-year phase-in of oversight requirements would “provide adequate time for laboratories and providers to be in compliance.”
The American Society of Clinical Oncology also put its weight behind the FDA framework.
“In contemporary oncology practice, a patient’s treatment options are increasingly driven by detection of molecular abnormalities in the tumor that drive treatment selection,” ASCO chief medical officer Richard L. Schilsky, MD, said in a Sept. 19 statement. “ASCO believes that the tests used to detect those abnormalities must be of the highest quality and thoroughly validated before being offered to doctors and patients. Our patients depend on high-quality tests as much as they depend on carefully studied, safe, and effective drugs to achieve the best possible outcomes.”
John L. Bishop, chairman and CEO of Sunnyvale, Calif.-based molecular diagnostics company Cepheid, says the FDA is headed in the right direction.
“Industry’s position has been that FDA should review novel and/or high-risk tests, regardless of who develops them, as a critical step to help assure test accuracy and patient safety,” he tells CAP TODAY. “I believe the new guidance addresses that concern by calling for review of the highest-risk tests soon after the guidance is finalized.
“Yet a gap between requirements for moderate-risk tests will remain in the near term,” Bishop adds. “Laboratories and manufacturers both can leverage other new guidances on benefit-risk assessments and balancing pre- and post-market requirements, working with the [FDA] to ensure that innovations and patient access to new technologies are not stifled, but encouraged. While other, more difficult details—such as whether and how laboratories comply with quality systems regulations—remain to be established, we commend the agency for its commitment to transparency, allowing for public input through the proposed public meeting and the extended 120-day comment period to the draft guidance. Open dialogue will allow all stakeholders to continue to explore oversight that strikes the appropriate balance between innovation, access, quality, and safety.”
Jeffrey Shuren, MD, JD, director of the FDA’s Center for Devices and Radiologic Health, spoke on behalf of the agency at the congressional hearing.
“We believe that LDTs serve an important role in health care and that there are many good tests on the market,” he said. “Unfortunately, FDA is also aware of faulty or unproven LDTs, including problems with several high-risk LDTs . . . FDA is concerned that people could initiate unnecessary treatment, or delay or forego treatment altogether, for a health condition, which could result in illness or death.”
Some in Congress have grumbled about the FDA’s plans. One is Rep. Michael C. Burgess, MD. He is a Texas Republican, ob-gyn, and vice chair of the Health Subcommittee that called the Sept. 9 hearing on LDTs. He said in a July 31 statement that “applying FDA’s regulatory approach to LDTs is redundant, will stifle innovation, and will require additional taxpayer funding for the FDA.” Burgess called instead for CLIA modernization.
Despite that and other concerns voiced about the FDA’s framework for LDTs, AdvaMedDx executive director Andrew Fish tells CAP TODAY that it is unlikely Congress will block the agency from taking action.
“I don’t rule out the prospect of some legislative effort to be more prescriptive to FDA on how it proceeds. But I’d still be surprised to see a meaningful legislative effort to prevent the FDA from moving forward,” says Fish. He testified at the Sept. 9 hearing in favor of the FDA framework on behalf of AdvaMedDx, which represents many mid-size and large diagnostic companies.
“One of the things I look to is not just the questions and the tone of the conversation in the hearing room,” Fish adds. “The bigger thing is the growing number of voices from different sectors . . . calling for the administration to move forward with this guidance. Payers are already moving ahead with scrutiny. There’s a strong majority of stakeholders saying that something needs to be done. This next phase of the debate will be very interesting. There will be the comment period, the public hearing, and an opportunity for all these players to comment much more, in greater detail, with another round of conversation on the complexities of how the FDA should move forward.”
Kevin B. O’Reilly is CAP TODAY senior editor.
What makes a high-risk LDT?
When deciding whether to require premarket review for laboratory-developed tests, the FDA has said it will consider:
- Whether the test is intended for use in high-risk diseases, conditions, or patient populations.
- Whether it is used for screening or diagnosis.
- The nature of the clinical decision made based on the test result.
- Whether a clinician or pathologist would have information beyond the LDT result to help guide diagnosis or treatment.
- What alternative testing and treatment options would be available to the patient.
- The potential consequences of erroneous results.
- The number and types of adverse events associated with the LDT.
Source: Food and Drug Administration. “Draft Guidance for Industry, Food and Drug Administration Staff, and Clinical Laboratories: Framework for Regulatory Oversight of Laboratory Developed Tests (LDTs).” Oct. 3, 2014.