
![]() CAP TODAY and the Association for Molecular Pathology have teamed up to bring molecular case reports to CAP TODAY readers. AMP members write the reports using clinical cases from their own practices that show molecular testing’s important role in diagnosis, prognosis, and treatment. The following report comes from Mayo Clinic, Rochester, Minn. If you would like to submit a case report, please send an email to the AMP at amp@amp.org. For more information about the AMP and all previously published case reports, visit www.amp.org.
CAP TODAY and the Association for Molecular Pathology have teamed up to bring molecular case reports to CAP TODAY readers. AMP members write the reports using clinical cases from their own practices that show molecular testing’s important role in diagnosis, prognosis, and treatment. The following report comes from Mayo Clinic, Rochester, Minn. If you would like to submit a case report, please send an email to the AMP at amp@amp.org. For more information about the AMP and all previously published case reports, visit www.amp.org.
Guang Yang, MD, PhD
David S. Viswanatha, MD
Rong He, MD
February 2020—Myeloproliferative neoplasms (MPNs) are clonal hematopoietic stem cell disorders characterized by the proliferation of one or more of the myeloid lineages. Philadelphia chromosome-negative classical MPNs include polycythemia vera (PV), primary myelofibrosis (PMF), and essential thrombocythemia (ET). The main recurrent driver mutations include Janus kinase 2 (JAK2) V617F, calreticulin (CALR), and myeloproliferative leukemia virus oncogene (MPL) mutations in PMF and ET, and JAK2 V617F and JAK2 exon 12 mutations in PV. Pathological diagnosis of MPNs follows the WHO classification, which provides specific major and minor criteria for each MPN subtype with incorporation of the molecular markers in the major criteria.1
Located on chromosome 19p13.2, the CALR gene encodes calreticulin (CALR), a multi-functional calcium-binding protein primarily involved in endoplasmic reticulum (ER) protein folding, calcium homeostasis, cellular proliferation, apoptosis, and immunogenic cell death.2 More than 50 types of pathogenic CALR mutations have been described in MPN. All were exon 9 (last exon) insertions and/or deletions (indels) resulting in a 1-bp frameshift leading to a specific alternative reading frame and a neoepitope in the carboxyl-terminus of the mutant CALR protein. The mutant CALR binds and constitutively activates MPL and the downstream JAK-STAT signaling pathway and leads to oncogenic transformation.2,3
Rarely, small in-frame CALR indels can be seen in healthy individuals or MPN patients. They are often germline polymorphisms, but they have also been reported as subclonal somatic events coexisting with classical MPN driver mutations in MPN patients. These findings indicate that in-frame CALR indels are of doubtful clinical significance, in contrast to frameshift indel alterations.2-6
As the 2016 WHO classification of myeloid neoplasms added CALR mutation into the major diagnostic criteria for PMF and ET, it is important for pathologists and clinicians to understand the genetic, biological, and clinical differences between the two types of CALR indels—frameshift versus in-frame—in clinical practice. Here we report an interesting case of an end-stage MPN patient harboring a pathogenic frameshift CALR mutation who successfully underwent stem cell transplantation with elimination of the neoplastic clone and acquisition of an in-frame CALR deletion from the healthy donor. This case illustrates the essential differences between the frameshift and in-frame indel variants in CALR.
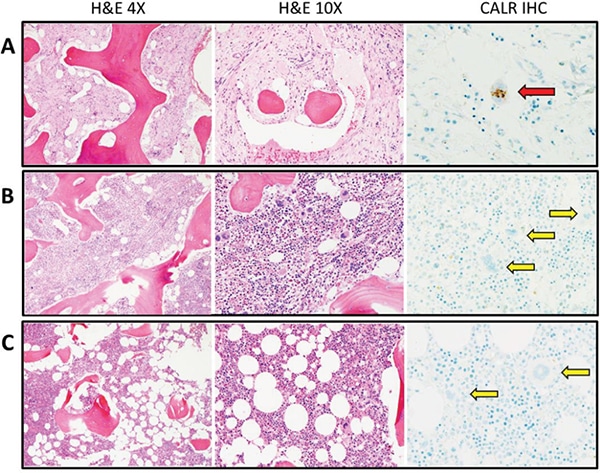
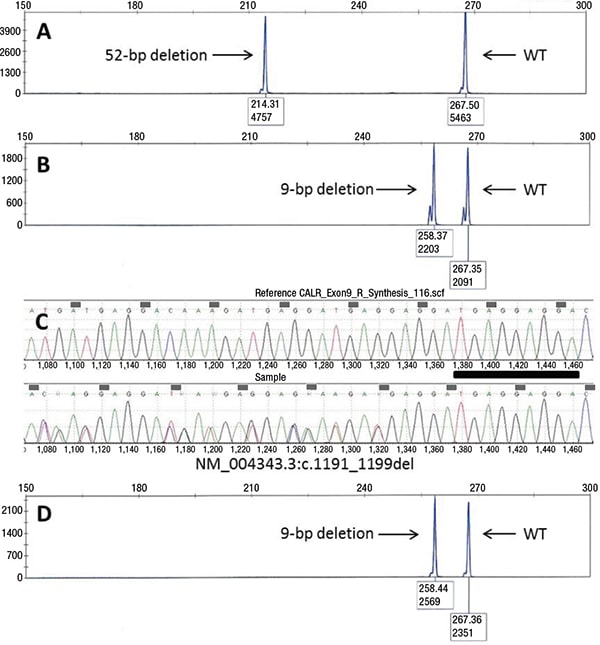
Case. A 42-year-old African American female was referred to our institution for further evaluation of progressive anemia, thrombocytopenia, and circulating blasts. She was diagnosed with ET 20 years ago at an outside institution with initial presenting platelet counts in the “one million range.” Past history was significant for intolerance to anagrelide, unprovoked deep vein thrombosis, and spontaneous miscarriages. Her CBC showed a hemoglobin of 8.9 g/dL, white blood cell count of 8.8 × 109/L, absolute neutrophil count of 1.3 × 109/L, and platelet count of 79 × 109/L. Peripheral blood smear showed a leukoerythroblastic picture with two percent metamyelocytes, 21 percent myelocytes, two percent promyelocytes, three percent circulating blasts, and six percent nucleated red blood cells. Dacrocytes were also present. The bone marrow aspirate was hemodilute, and the core biopsy demonstrated a markedly hypocellular marrow with marked panhypoplasia, grade 3/3 myelofibrosis with collagen fibrosis, osteosclerosis, and dilated sinusoids. Rare residual megakaryocytes were present and stained positive by the monoclonal CAL2 antibody, which specifically recognizes the C-terminal neoepitope of the mutant CALR proteins resulting from the 1-bp frameshift pathogenic CALR mutations (Fig. 1A).7 Molecular testing was negative for JAK2 V617F and MPL exon 10 mutations, whereas a frameshift 52-bp deletion was detected in CALR exon 9 by capillary electrophoresis fragment sizing following polymerase chain reaction using primers flanking exon 9 of CALR (Fig. 2A).
Chromosome analysis showed del(12p) and del(13q) in 20/20 metaphases. Lactate dehydrogenase was elevated at 531 U/L.
The overall findings, including the documented previous diagnosis of ET and the long duration between the initial ET diagnosis and myelofibrosis development, presence of a pathogenic CALR mutation, bone marrow morphology and myelofibrosis, peripheral blood leukoerythroblastic picture, anemia, and elevated LDH, were compatible with a diagnosis of post-ET myelofibrosis per the 2016 WHO diagnostic criteria.1 A repeat bone marrow examination two months later showed similar morphologic findings; however, the CBC showed significantly decreased Hb, ANC, and Plt, with 13 percent circulating blasts and 17 percent blasts in a hemodilute bone marrow aspirate. The patient received cytoreductive chemotherapy with idarubicin and cytarabine, followed by myeloablative conditioning with busulfan and cyclophosphamide. She subsequently underwent peripheral blood stem cell transplant from a matched healthy sibling donor two months later.
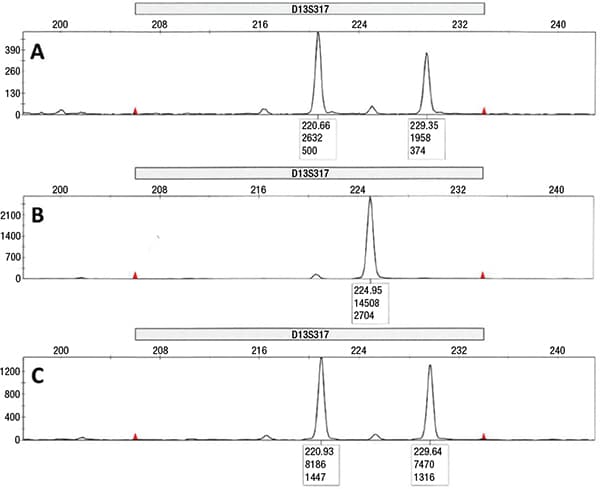
Three months after the transplant, the patient’s CBC improved to a Hb of 9.3 g/dL, WBC 4.9 × 109/L, ANC 2.9 × 109/L, and Plt 87 × 109/L. Bone marrow examination exhibited a spectrum of changes, with patchy areas showing marked hypocellularity in a fibrotic background similar to the pre-transplant bone marrow, and other areas exhibiting cellular reconstitution with trilineage hyperplasia and cellular streaming. The megakaryocytes did not show distinct tight clustering, although focal loose aggregates were noted; they were negative for mutant CALR by the CAL2 immunostain (Fig. 1B). Background osteosclerosis was still present. Chromosome analysis showed a normal karyotype. Repeat CALR mutational analysis revealed disappearance of the frameshift 52-bp deletion and, interestingly, acquisition of an in-frame 9-bp deletion in CALR (Fig. 2B). The sample was forwarded for Sanger sequencing, which confirmed the presence of a 9-bp deletion (NM_004343.3:c.1191_1199del; p.Glu398_Asp400del, Fig. 2C). Concurrent chimerism testing showed 100 percent donor and zero percent recipient DNA (Fig. 3). Retrospective CALR testing on the donor DNA was performed and further confirmed the donor-origin of the 9-bp deletion (Fig. 2D).
A follow-up examination nine months later showed further improvement of CBC counts: Hb 11.0 g/dL, WBC 4.6 × 109/L, ANC 3.9 × 109/L, and Plt 89 × 109/L. Bone marrow examination was normocellular with unremarkable trilineage hematopoiesis, and no morphologic features of involvement by an MPN. Megakaryocytes showed no clustering and were negative by CAL2 immunostain (Fig. 1C). Molecular testing again revealed 100 percent donor DNA, zero percent recipient DNA, and persistence of the in-frame 9-bp deletion. Karyotype was also normal. At the last follow-up, 31 months after the peripheral blood stem cell transplant, the patient showed no sign of recurrence of an MPN.
Discussion. The Philadelphia chromosome-negative classical MPNs ET, PV, and PMF are chronic myeloid neoplasms with significant risks for leukemic transformation and terminal (end stage) myelofibrosis. Decades of research have greatly facilitated our understanding of the genetic basis of MPN pathogenesis and largely defined their mutational landscape by recurrent driver mutations in JAK2, CALR, and MPL, in an essentially mutually exclusive pattern. In ET and PMF, the most common mutation is the missense mutation JAK2 V617F, seen in 50 percent to 60 percent of patients, followed by CALR exon 9 mutation (20 to 30 percent), and MPL exon 10 mutation (five to 10 percent in PMF and three to five percent in ET). In PV, at least 95 percent of patients harbor a JAK2 V617F, and the vast majority of the remainder has JAK2 exon 12 mutations.8 These mutations converge on the activation of the JAK-STAT pathway, which also explains their mutually exclusive nature and the observed clinical response to JAK2 inhibitors in MPN patients regardless of the specific driver mutation.9
Although JAK2 V617F and MPL mutations were documented in MPN more than a decade ago, the discovery of CALR in MPN was more recent. In 2013, two European groups independently reported highly prevalent somatic mutations in the last exon of CALR in the majority of JAK2 and MPL-negative ET and PMF patients but not PV patients.2,3 CALR mutations identified were exclusively insertions and/or deletions resulting in a 1-bp frameshift following the stereotypical pattern of (3n+1) bp for deletions and (3n+2) bp for insertions. The two most frequent mutations were the 52-bp deletion p.L367fs*46 (type 1) and 5-bp insertion p.K385fs*47 (type 2), and they account for approximately 53 percent and 32 percent of the CALR-mutated MPN cases, respectively. These pathogenic frameshift mutations result in a specific alternative reading frame and generate a C-terminal neomorphic mutant CALR protein. CALR frameshift mutations have been found in the neoplastic hematopoietic stem and progenitor cells in MPN. Studies have shown that these mutations lead to constitutive signaling through the JAK-STAT pathway by direct activation of thrombopoietin receptor MPL, independent of thrombopoietin ligand activation. The C-terminal neoepitope of mutant CALR is indispensable for the activation of MPL and presumptively mediates the pivotal interaction through induction of CALR conformational change. In a retroviral mouse model, CALR mutants were sufficient to induce an MPN phenotype mimicking ET with later progression to myelofibrosis.10-12
Clinically, in cases morphologically equivocal for MPN and/or with unexplained cytosis or myelofibrosis, identification of a driver mutation aids in establishing a diagnosis of MPN in the appropriate clinical setting. The mutational status of JAK2, MPL, and CALR is also prognostically informative in PMF because a triple-negative (JAK2/CALR/MPL-wild type) mutational status is an adverse molecular signature in PMF, and type 1/like CALR mutations have been shown to be associated with more favorable clinical outcome. In ET, CALR mutations are associated with a lower risk of thrombosis.2,3 In light of this apparent clinical and pathologic validity, the 2016 WHO classification of myeloid neoplasms listed CALR along with JAK2 and MPL as one of the major diagnostic criterion for ET and PMF.1
Interestingly but not surprisingly, besides the pathogenic frameshift mutations seen in MPN, rare small in-frame (3n) indels (≤ 18-bp) of CALR have been described in both healthy individuals and in MPN patients. These are commonly germline polymorphisms but can also occur as somatic events. The Klampfl study reported three cases with in-frame CALR indels: 1/524 healthy individuals with a 3-bp deletion, and 2/292 JAK2 V617F-positive PMF and ET patients with germline CALR deletions (an 18-bp deletion in 1/108 PMF and a 12-bp deletion in 1/184 ET).3 Nangalia, et al., also reported infrequent germline 9-bp and 12-bp CALR deletions confirmed through mining of public databases, exome sequencing of healthy donors, and paired constitutional T-cell population sequencing.2 The findings were further supported by subsequent reports of approximately one percent (8/809) cases suspicious/diagnosed of MPN showing a small in-frame CALR deletion of 3- or 9-bp, all at a variant allele fraction of about 50 percent, consistent with germline origin.5 Two cases co-harbored classical MPN driver mutations, with MPL W515L in one and JAK2 V617F in the other. The p.Glu398_Asp400del 9-bp deletion seen in our patient has been documented in the public databases as a low-level polymorphism with an overall population frequency of 0.13 percent, and variable frequencies in different ethnicity groups, ranging from zero percent in the Finnish Europeans up to 1.3 percent in the African population.13
In addition to germline polymorphic variants, CALR in-frame deletions may also present as a somatic event in MPN. This was first documented as a rare case of subclonal acquisition of an 18-bp CALR deletion on the same allele of a precedent 1-bp CALR frameshift deletion in an MPN patient; in this case, the specific mutant reading frame and the C-terminal neoepitope of the frameshift mutation was retained.3 Lim, et al., also reported four types of 3-bp CALR deletions in 5/59 JAK2 V617F-positive ET patients using TA cloning following negative Sanger sequencing. The somatic nature of these 3-bp deletions was supported by their low allele burden levels (1.4 percent to 10 percent).4 In both studies, these subclonal somatic in-frame deletions were of doubtful clinical significance and most likely represented passenger events, in light of the presence of coexisting classical MPN driver mutations in these MPN cases. Although Lim, et al., showed a higher frequency of CALR in-frame deletion in JAK2 V617F-mutated MPN, it is worth noting that the relatively small study cohort and the sensitive technique of TA cloning used in the study may have contributed to the differences seen between studies. Biologically, it is evident that these in-frame indels produce neither a reading frameshift nor the C-terminal neoepitope indispensable for the oncogenic activation of MPL in the pathogenesis of mutant CALR MPN. In-frame CALR indels are therefore of doubtful clinical significance. Unfortunately, the fundamental differences between the frameshift and in-frame CALR indels are sometimes underappreciated clinically, leading to erroneous molecular pathologic interpretation of the latter.6
In the current case, the patient presented with post-ET myelofibrosis with blast transformation. She harbored a frameshift 52-bp deletion in CALR, conforming to the stereotypic (3n+1) bp pattern of pathogenic CALR deletions. She was also negative for JAK2 V617F and MPL mutations, consistent with the observed mutually exclusive nature of the three MPN driver mutations. After a successful peripheral blood stem cell transplant, the patient’s frameshift 52-bp deletion was eliminated, but interestingly an in-frame 9-bp deletion in CALR appeared. We confirmed the donor origin of this in-frame CALR deletion through both concurrent chimerism testing, which demonstrated 100 percent donor and zero percent recipient, and retrospective confirmation of the presence of the 9-bp in-frame deletion in the donor sample. The patient remained disease-free at the last follow-up 31 months status post-transplant. This case showcases the genetic, biologic, and clinical differences between a frameshift CALR mutation with established pathogenic role in MPN and an in-frame CALR deletion of doubtful clinical significance acquired from a healthy donor. It would be important for practicing pathologists and clinicians to recognize the salient differences between these two types of CALR indel variants to avoid errant interpretation and subsequent misdiagnosis.
The commonly used methods for CALR mutational analysis are Sanger sequencing and next-generation sequencing, the latter generally providing better analytical sensitivity. As the pathogenic CALR mutations in MPN are indels, they are also readily detected by fluorescent capillary electrophoresis fragment sizing analysis. One caveat is that the accuracy of capillary electrophoresis fragment sizing is usually ±1–3-bp depending on the size of the PCR products and the platform used, requiring rigorous assay validation to ensure indel sizing accuracy. For rare CALR indels with sizes not covered by validated test run controls, supplemental sequencing is recommended to confirm the exact sizes and accurately differentiate between a frameshift versus in-frame CALR indel. In the case presented, as our test runs included positive controls of the most common 52-bp deletion and 5-bp insertion, the 9-bp deletion seen by fragment analysis was further Sanger sequenced for size confirmation. Additionally, as the pathogenic frameshift CALR mutations generate a C-terminal neoepitope, several antibodies have been successfully developed against this novel peptide region and validated for clinical immunohistochemical use. The immunostain may thus serve as a surrogate marker for the presence of pathogenic frameshift CALR mutations and provides a convenient alternative for molecular CALR testing in situations where the latter test is not readily available.7,14
Another pertinent observation is that in diseases with pathologic alterations in bone marrow microenvironment (for example, MPN exhibiting myelofibrosis and osteosclerosis), molecular marker tracking may provide a more accurate reflection of therapeutic response than morphologic evaluation, particularly in the early stage after stem cell transplantation, given the delay for the engrafted cells to modify and reconstruct the microenvironment. In our case, although the patient lost the pathogenic frameshift 52-bp CALR deletion three months after peripheral blood stem cell transplant and showed 100 percent donor DNA by chimerism analysis, the bone marrow findings provided no clear morphologic evidence of disease resolution. The abnormal MPN-like morphological findings eventually subsided in the subsequent bone marrow biopsy nine months later, lagging behind the molecular finding.
Conclusion. Here we present a rare case of a post-ET myelofibrosis patient harboring a pathogenic frameshift CALR mutation who underwent a successful peripheral blood stem cell transplantation. Along with clearance of her neoplastic frameshift mutation, she acquired an in-frame CALR deletion from the healthy donor. This case illustrates the salient differences between the pathogenic frameshift CALR mutations and the in-frame CALR indels of doubtful clinical significance. It also emphasizes the importance for pathologists and clinicians to recognize these differences to avoid diagnostic errors and mismanagement in clinical practice.
- Orazi A, Bennett JM, Germing U, et al. Chronic myelomonocytic leukaemia. In: Swerdlow SH, Campo E, Harris NL, et al., eds. WHO Classification of Tumours of Haematopoietic and Lymphoid Tissues; vol 2. Rev. 4th ed. Lyon, France: IARC Press; 2017:82.
- Nangalia J, Massie CE, Baxter EJ, et al. Somatic CALR mutations in myeloproliferative neoplasms with nonmutated JAK2. N Engl J Med. 2013;369(25):2391–2405.
- Klampfl T, Gisslinger H, Harutyunyan AS, et al. Somatic mutations of calreticulin in myeloproliferative neoplasms. N Engl J Med. 2013;369(25):2379–2390.
- Lim KH, Chang YC, Gon-Shen Chen C, et al. Frequent CALR exon 9 alterations in JAK2 V617F-mutated essential thrombocythemia detected by high-resolution melting analysis. Blood Cancer J. 2015;5:e295.
- Wang Y, Ho AK, Pan Q, Racke FK, Jones D. In-frame exon 9 CALR deletions co-occur with other alterations in the JAK-STAT pathway in myeloproliferative neoplasms. Blood. 2014;124(21):4588.
- He R, Hanson CA, Chen D, et al. Not all CALR mutations are created equal. Leuk Lymphoma. 2015;56(8):2482–2483.
- Stein H, Bob R, Dürkop H, et al. A new monoclonal antibody (CAL2) detects CALRETICULIN mutations in formalin-fixed and paraffin-embedded bone marrow biopsies. Leukemia. 2016;30(1):131–135.
- Scott LM, Tong W, Levine RL, et al. JAK2 exon 12 mutations in polycythemia vera and idiopathic erythrocytosis. N Engl J Med. 2007;356(5):459–468.
- Nangalia J, Green AR. Myeloproliferative neoplasms: from origins to outcomes. Hematology Am Soc Hematol Educ Program. 2017;2017(1):470–479.
- Araki M, Yang Y, Masubuchi N, et al. Activation of the thrombopoietin receptor by mutant calreticulin in CALR-mutant myeloproliferative neoplasms. Blood. 2016;127(10):1307–1316.
- Araki M, Komatsu N. Novel molecular mechanism of cellular transformation by a mutant molecular chaperone in myeloproliferative neoplasms. Cancer Sci. 2017;108(10):1907–1912.
- Chachoua I, Pecquet C, El-Khoury M, et al. Thrombopoietin receptor activation by myeloproliferative neoplasm associated calreticulin mutants. Blood. 2016;127(10):1325–1335.
- Karczewski KJ, Francioli LC, Tiao G, et al. Variation across 141,456 human exomes and genomes reveals the spectrum of loss-of-function intolerance across human protein-coding genes. BioRxiv website. doi:10.1101/531210. Originally posted Jan. 28, 2019.
- Vannucchi AM, Rotunno G, Bartalucci N, et al. Calreticulin mutation-specific immunostaining in myeloproliferative neoplasms: pathogenetic insight and diagnostic value. Leukemia. 2014;28(9):1811–1818.
Dr. Yang is a molecular genetic pathology fellow, Division of Laboratory Genetics and Genomics; Dr. Viswanatha is an associate professor, co-director of the molecular hematopathology laboratory and clinical genome sequencing laboratory, Division of Hematopathology and Division of Laboratory Genetics and Genomics; Dr. He is an associate professor, co-director of the molecular hematopathology laboratory, Division of Hematopathology—all in the Department of Laboratory Medicine and Pathology, Mayo Clinic, Rochester, Minn.
Test yourself
[tabs type=”horizontal”][tabs_head][tab_title]Here are three questions taken from the case report.[/tab_title][tab_title]Answers[/tab_title][/tabs_head][tab]Answers are online at www.amp.org/casereports and will be published next month in CAP TODAY.
1. Which of the following statements is false about driver mutations in Philadelphia chromosome-negative myeloproliferative neoplasms?
a. The most common driver mutation is JAK2 V617F.
b. CALR exon 9 mutations are the second most common mutations in polycythemia vera.
c. MPL exon 10 mutations can be seen in primary myelofibrosis.
d. JAK2 V617F, CALR, and MPL mutations are essentially mutually exclusive of each other in essential thrombocythemia.
2. In the workup of a myeloproliferative neoplasm, which of the following CALR genetic alterations does not provide supporting evidence of involvement by an MPN?
a. 52 base pair deletion
b. 5 base pair insertion
c. 12 base pair deletion
d. 34 base pair deletion
3. Which one of the following statements is true about CALR?
a. It is located on chromosome 18.
b. The two most common CALR mutations in myeloproliferative neoplasms are 52-bp deletion and 5-bp insertion.
c. CALR mutations in myeloproliferative neoplasms lead to inactivation of signaling through the JAK-STAT pathway.
d. In-frame insertion/deletions of CALR are common pathogenic mutations seen in myeloproliferative neoplasms.
[/tab]
[tab]
1. Which of the following statements is false about driver mutations in Philadelphia chromosome-negative myeloproliferative neoplasms?
a. The most common driver mutation is JAK2 V617F.
b. CALR exon 9 mutations are the second most common mutations in polycythemia vera.
c. MPL exon 10 mutations can be seen in primary myelofibrosis.
d. JAK2 V617F, CALR, and MPL mutations are essentially mutually exclusive of each other in essential thrombocythemia.
2. In the workup of a myeloproliferative neoplasm, which of the following CALR genetic alterations does not provide supporting evidence of involvement by an MPN?
a. 52 base pair deletion
b. 5 base pair insertion
c. 12 base pair deletion
d. 34 base pair deletion
3. Which one of the following statements is true about CALR?
a. It is located on chromosome 18.
b. The two most common CALR mutations in myeloproliferative neoplasms are 52-bp deletion and 5-bp insertion.
c. CALR mutations in myeloproliferative neoplasms lead to inactivation of signaling through the JAK-STAT pathway.
d. In-frame insertion/deletions of CALR are common pathogenic mutations seen in myeloproliferative neoplasms.
[/tab]
[/tabs]