
CAP TODAY and the Association for Molecular Pathology have teamed up to bring molecular case reports to CAP TODAY readers. AMP members write the reports using clinical cases from their own practices that show molecular testing’s important role in diagnosis, prognosis, and treatment. The following report comes from the University of New Mexico. If you would like to submit a case report, please send an email to the AMP at amp@amp.org. For more information about the AMP and all previously published case reports, visit www.amp.org.
Brittany Coffman, MD
Brian Menkhaus, MD
Devon Chabot-Richards, MD
![]() April 2019—Acute promyelocytic leukemia (APL) is a subtype of acute myeloid leukemia (AML) in which promyelocytes predominate. APL accounts for about 10 percent of AML cases, and although APL can be diagnosed at any age, it is most common among young adults with a slight male predominance.1 APL is defined by the balanced reciprocal translocation (15;17)(q22;q21) between PML and RARA, although variant translocations involving RARA and other partner genes can occur. The fusion results in uncontrolled cell proliferation and inhibition of cell differentiation.
April 2019—Acute promyelocytic leukemia (APL) is a subtype of acute myeloid leukemia (AML) in which promyelocytes predominate. APL accounts for about 10 percent of AML cases, and although APL can be diagnosed at any age, it is most common among young adults with a slight male predominance.1 APL is defined by the balanced reciprocal translocation (15;17)(q22;q21) between PML and RARA, although variant translocations involving RARA and other partner genes can occur. The fusion results in uncontrolled cell proliferation and inhibition of cell differentiation.
The PML gene, or promyelocytic leukemia gene, is located on chromosome 15.2 Its gene product is a tumor suppressor protein that blocks cell proliferation and regulates apoptosis via FAS ligand and tumor necrosis factor-alpha. The RARA gene, or retinoic acid receptor-alpha, is located on chromosome 17.3 The RARA gene codes for a nuclear receptor that regulates gene transcription, including genes involved in differentiation, apoptosis, and granulopoiesis. The translocation between PML and RARA occurs between the long arms of both chromosomes 15 and 17, respectively. The breakpoint of RARA occurs at intron 2, but the breakpoint of PML can occur at three different locations, resulting in different transcript sizes with similar functions (Fig. 1).4
The most common breakpoint subtype of PML, bcr-1 (breakpoint cluster region) or long form, is at intron 6, which occurs in 45 to 55 percent of APL cases.4 The second most common is the short form, or bcr-3, which occurs in 35 to 45 percent of cases and occurs at intron 3 of PML. Finally, the least common subtype is the variable form, or bcr-2, which occurs in five to 10 percent of cases and occurs at exon 6 of PML. The bcr-2 form is termed variable form because the breakpoints can occur at different sites within exon 6. The resulting PML-RARA fusion gene product prevents normal transcription, which ultimately leads to lack of differentiation of myeloid cells and provides cells with a survival advantage.

Patients may present with nonspecific symptoms such as fever, fatigue, decreased appetite, and/or weight loss; however, a subset may also present with bleeding due to disseminated intravascular coagulation (DIC), which is associated with APL.5 Rapid diagnosis is imperative in cases of APL due to the increased risk of death secondary to DIC in this patient population. Most patients demonstrate pancytopenia on complete blood count, though some patients present with leukocytosis. Leukocytosis is more common in the microgranular variant of APL.
Two morphologic forms of APL are recognized including hypergranular (or typical) and microgranular variants. The blasts of typical APL have irregular large nuclei that can be bilobed and may have a “sliding plate” morphology. The cytoplasm is densely packed with large granules and occasional Auer rods. Microgranular variant blasts mostly have a bilobed morphology, and cytoplasmic granules are not apparent as they are submicroscopic. Cytochemical stains such as myeloperoxidase and Sudan black are strongly positive in both variants, often to the point that the cytoplasmic granules obscure the nucleus.
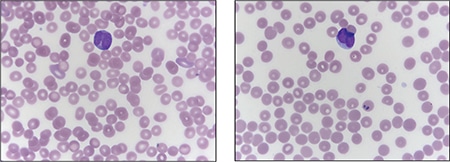
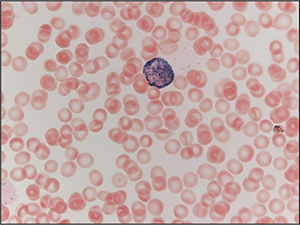
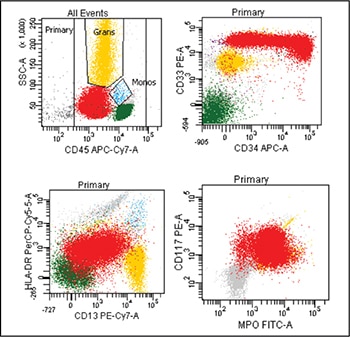
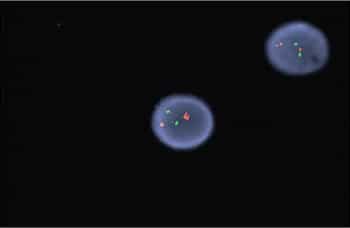
Case. A 16-year-old male presented with a one-week history of fatigue and easy bruising. Complete blood count revealed a white count of 4.8 × 103/mm3 with left shift of the myeloid lineage with 50 percent blasts, hematocrit of 31 percent, hemoglobin of 10.8 g/dL, and a platelet count of 29 × 103/mm3. Review of the peripheral blood smear showed numerous blasts with increased nuclear-cytoplasmic ratio and bilobed nuclei with sliding plate morphology (Fig. 2). Myeloperoxidase stain performed on the peripheral blood was strongly positive (Fig. 3). The morphologic and clinical findings were concerning for APL and thus the clinical team started ATRA (all-trans-retinoic acid) while awaiting final diagnosis. Flow cytometry revealed the blast population to be dim CD45, bright CD33, subset CD34, dim HLA-DR, dim CD13, CD117, subset CD56, and cytoplasmic MPO positive (Fig. 4). Dual color dual fusion fluorescence in situ hybridization was negative for t(15;17) (Fig. 5).6
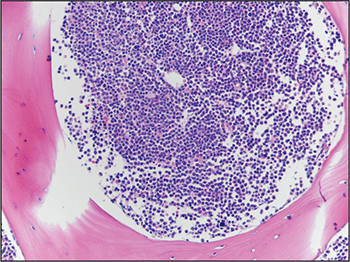
A bone marrow biopsy was then performed and revealed a hypercellular marrow with left shifted maturation in the myeloid lineage with 77 percent blasts (Fig. 6). Blast morphology was similar to the peripheral blood. Flow cytometry performed on the bone marrow was identical to the peripheral blood. Conventional karyotype performed on the bone marrow revealed a normal male chromosome complement with no abnormalities identified. Due to the clinical suspicion of APL along with blast morphology and immunophenotype, despite negative FISH and karyotype, reverse transcription-polymerase chain reaction (RT-PCR) testing was obtained.7 Testing revealed a cryptic t(15;17) with the bcr-3 transcript, and the patient was formally diagnosed with APL (Fig. 7). Despite starting ATRA, the patient developed DIC, which was effectively treated with transfusion. The patient was discharged home one month later and remains in remission more than three years later.
Discussion. APL is an acute leukemia that is defined by t(15;17) and requires prompt diagnosis because it is associated with DIC. Review of blast morphology and cytochemical stains can aid the pathologist in narrowing the differential diagnosis, but the final diagnosis of APL often requires additional testing. It is common for rapid turnaround FISH to be performed to identify the t(15;17), thus securing the diagnosis. However, in cases such as this one, FISH and karyotype will fail to identify the cryptic translocation. Delay in diagnosis in these cases may be detrimental and even lead to death if ATRA therapy is not begun.
Cryptic translocations of APL account for about two percent to four percent of all APL cases. The majority of cryptic translocations result from submicroscopic insertions of RARA into PML, but complex rearrangements involving numerous chromosomes can also be a cause. In addition, variant translocations between RARA and other genes can occur. Variant translocations may require additional FISH probes as they may not be recognized by usual t(15;17) dual color probes; it is important to recognize, however, that these are not cryptic translocations but variant translocations. Furthermore, all cryptic and variant alterations involve RARA but may not involve PML, highlighting the importance of RARA gene alterations in the development of APL.
Of note, cases of variant translocations involving RARA but not PML may have blasts morphologically resembling APL but with a worse prognosis, specifically t(11;17)(q23;q21), which may also be resistant to ATRA. These cases are best classified as APL with a variant RARA translocation per the 2016 WHO.8 The majority of cryptic cases of APL have similar morphology, immunophenotype, and cytochemical staining patterns, which should raise clinical concern for APL. In these cases, RT-PCR, microarray, and gene sequencing can identify the cryptic translocation and definitively establish the proper diagnosis.
Although it may seem that the initial best option would be to use RT-PCR testing primarily, each testing modality has its own advantages. FISH carries the advantage of there being specific probes to detect the translocation and can often be completed within 24 hours. In addition, break-apart probes can be used to identify fusions with other partners, an advantage when dealing with variant translocations. The disadvantages of FISH are its insensitivity for minimal residual disease monitoring and the possibility of missing cryptic cases, such as this case. Conventional karyotype, with a longer turnaround time of five to 10 days, can identify the usual t(15;17) and may also identify other genetic abnormalities that have prognostic significance. Karyotype may also miss cryptic translocations and it too is insensitive to monitoring for minimal residual disease. RT-PCR is by far the most sensitive testing modality and is frequently used for minimal residual disease monitoring and to identify cryptic translocations. Turnaround time, however, is typically longer than for FISH. RT-PCR has the added advantage that it can identify the specific translocation subtype, including bcr-1, -2, or -3.
A final option for identifying genetic abnormalities in such cases is next-generation sequencing. It is by far the most sensitive method of identifying genetic abnormalities because it can identify numerous abnormalities, not limited to cryptic translocations.9 Historically, the clinical use of next-generation sequencing has been limited due to high cost and long turnaround time; however, with technology advances the cost and turnaround time have been significantly decreased. Even with these advances, next-generation sequencing is not readily available in every laboratory.
In cases where APL is suspected it is important to obtain rapid turnaround FISH and to alert the clinical team of the suspected diagnosis so ATRA therapy can be initiated promptly to prevent DIC. If FISH is demonstrated to be negative in a case with typical APL morphology and immunophenotype, it is important to perform additional molecular testing to rule out cryptic translocation before assigning a diagnosis other than APL. With appropriate diagnosis and treatment, patients with APL have an excellent prognosis.
- Paulson K, Serebrin A, Lambert P, et al. Acute promyelocytic leukaemia is characterized by stable incidence and improved survival that is restricted to patients managed in leukaemia referral centres: a pan-Canadian epidemiological study. Br J Haematol. 2014;166(5):660–666.
- Salomoni P, Dvorkina M, Michod D. Role of the promyelocytic leukaemia protein in cell death regulation. Cell Death Dis. 2012;3:e247.
- Pandolfi PP. Oncogenes and tumor suppressors in the molecular pathogenesis of acute promyelocytic leukemia. Hum Mol Genet. 2001;10(7):769–775.
- Choppa PC, Gomez J, Vall HG, Owens M, Rappaport H, Lopategui JR. A novel method for the detection, quantitation, and breakpoint cluster region determination of t(15;17) fusion transcripts using a one-step real-time multiplex RT-PCR. Am J Clin Pathol. 2003;119(1):137–144.
- Adams J, Nassiri M. Acute promyelocytic leukemia: a review and discussion of variant translocations. Arch Pathol Lab Med. 2015;139(10):1308–1313.
- Campbell LJ, Oei P, Brookwell R, et al. FISH detection of PML-RARA fusion in ins(15;17) acute promyelocytic leukaemia depends on probe size. Biomed Res Int. Epub March 28, 2013. doi:10.1155/2013/164501.
- Gabert J, Beillard E, van der Velden VH, et al. Standardization and quality control studies of ‘real-time’ quantitative reverse transcriptase polymerase chain reaction of fusion gene transcripts for residual disease detection in leukemia—a Europe Against Cancer program. Leukemia. 2003;17(12):2318–2357.
- Swerdlow SH, Campo E, Harris NL, et al., eds. WHO Classification of Tumours of Haematopoietic and Lymphoid Tissues. Vol 2. Revised 4th ed. Lyon, France: WHO Press; 2017.
- Welch JS, Westervelt P, Ding L, et al. Use of whole-genome sequencing to diagnose a cryptic fusion oncogene. JAMA. 2011;305(15):1577–1584.
Dr. Coffman is a pathology resident and Dr. Chabot-Richards is associate professor of hematopathology and molecular pathology—both in the Department of Pathology, University of New Mexico, Albuquerque. Dr. Chabot-Richards is also medical director, TriCore Reference Laboratories, Albuquerque. Dr. Menkhaus, formerly a pathology resident at UNM, is in the Department of Pathology, Memorial Hospital of Sheridan County, Sheridan, Wyo.
Test yourself: Here are three questions taken from the case report.
Answers are online now at www.amp.org/casereports and will be published next month in CAP TODAY.
1. Which of the following is true regarding acute promyelocytic leukemia?
a. Patients are always older.
b. Patients may present in DIC.
c. Diagnosis requires identification of t(8;21).
d. Patients always present with leukocytosis.
2. The long form, or bcr-1, breakpoint accounts for what percentage of acute promyelocytic leukemia cases?
a. 15–20 percent
b. 80–90 percent
c. 45–55 percent
d. 35–45 percent
3. What is an advantage of performing fluorescence in situ hybridization over reverse transcription-polymerase chain reaction?
a. Rapid turnaround time
b. Ability to detect cryptic translocations
c. More sensitive for minimal residual disease
d. Allows for subtyping of breakpoint
Test yourself answers for March 2019 case report
In the March 2019 issue was a case report, “FDA-approved DNA blood test for colorectal cancer prompts patient to undergo colonoscopy,” written by members of the Association for Molecular Pathology. Here are answers (in bold) to the three “test yourself ” questions that followed that case report.
1. The five-year survival for patients diagnosed with stage I CRC is closest to:
a. 10 percent
b. 35 percent
c. 50 percent
d. 90 percent
2. In the United States, the number of age-eligible individuals who are not compliant with guideline-recommended CRC screening is closest to:
a. 1 million
b. 1 million
c. 20 million
d. 100 million
3. The FDA-approved SEPT9 test is indicated for:
a. Symptomatic patients who refuse other CRC screening methods.
b. Postoperative surveillance in patients with stage II CRC.
c. Asymptomatic, average-risk, age-eligible patients who have refused other screening methods.
d. Predictive testing in asymptomatic patients with a family history of CRC.