
Amy Carpenter
May 2025—How the World Health Organization fifth edition of hematolymphoid tumors and the International Consensus Classification differ for myeloid malignancies was highlighted in cases presented in a CAP24 session last fall.
Second of two parts
Part one in the April issue
Sanam Loghavi, MD, associate professor of pathology and laboratory medicine, Department of Hematopathology, University of Texas MD Anderson Cancer Center, spoke of myelodysplastic neoplasms/syndromes (MDS) with defining genetic abnormalities and the allelic state of TP53 in MDS, among other things. (Kamran M. Mirza, MD, PhD, of the University of Michigan, co-presented. See part one in the April issue: https://bit.ly/CT_0425-myeloid.)
Dr. Loghavi’s first case was that of a 51-year-old woman who underwent a workup for pancytopenia. The patient’s CBC showed absolute neutropenia with a low white blood cell count (1.2 × 109/L). She had anemia (Hgb: 9.8 g/dL) and thrombocytopenia (Plt: 41 × 109/L). Her MCV was normal (92 fL).
The bone marrow biopsy (Fig. 1) was fibrotic and hypercellular for age, Dr. Loghavi said. The reticulin stain (middle) confirmed the presence of fibrosis. Often in cases that have high degrees of fibrosis, “the aspirate smears are terrible. You cannot see dysplasia; it’s difficult to locate the blasts” without looking at many aspirate smears, which is what they did. Two blasts can be seen on the aspirate smear (right). “Overall there were 16 percent blasts but in a hemodiluted smear,” she said. (More on this case later.)
In the WHO fifth edition (WHO-HEM5) in general, “the genetic categories trump morphologic categories,” Dr. Loghavi said (Khoury JD, et al. Leukemia. 2022;36[7]:1703–1719).
One such genetic category is MDS with low blasts and isolated 5q deletion, which she calls “a misnomer because despite what the terminology implies, you can have one additional set of genetic abnormalities as long as it doesn’t involve chromosome 7,” because the latter is associated with a poor prognosis and del(5q) is typically thought to be a lower-risk disease. (In the WHO fifth edition, myelodysplastic syndromes were renamed myelodysplastic neoplasms—abbreviated MDS.)
Another genetically defined category of MDS in the WHO classification is MDS with low blasts and SF3B1 mutation (MDS-SF3B1). The variant allelic frequency qualifier is one difference between the MDS with SF3B1 mutation categories in the WHO and ICC classifications (five percent, WHO; 10 percent, ICC).
The WHO allows for a branching category of MDS with SF3B1 mutation, Dr. Loghavi said. “If you don’t have a mutation or are in a resource-poor setting without [access to] sequencing and you have more than 15 percent ring sideroblasts, you can still put the case in this diagnostic category, but instead of calling it ‘with SF3B1 mutation,’ you call it MDS with ring sideroblasts.” The reason: In the initial trials studying response to luspatercept [Reblozyl], patients with ring sideroblasts and SF3B1 mutations benefited the most from the drug. “For the ICC,” Dr. Loghavi said, “you need to have the mutation and only that is recognized as a distinct category.”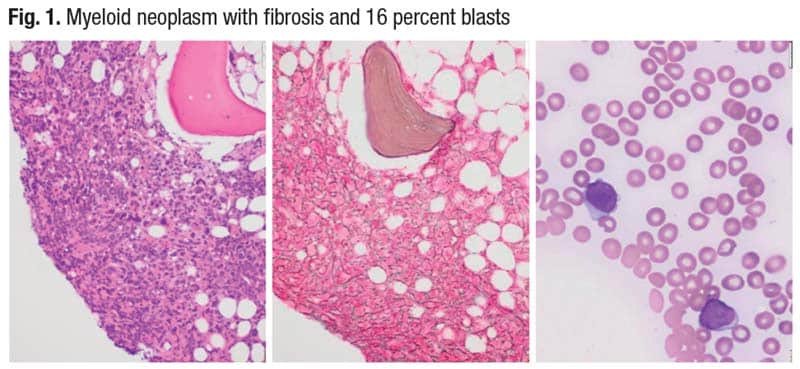
The third category of MDS with defining genetic abnormalities is MDS with biallelic TP53 inactivation (for now, it is not AML-defining in WHO). Its defining features in WHO are less than 20 percent blasts in bone marrow and peripheral blood, usually complex cytogenetics, and two or more TP53 mutations or one mutation with evidence of TP53 copy number loss or copy neutral loss of heterozygosity. The WHO uses the term biallelic, and the ICC uses multi-hit, “which essentially means you lose both functional copies of TP53 by different means,” Dr. Loghavi said. It’s her view that “the ICC approach is probably more accurate in this case” (Arber DA, et al. Blood. 2022;140[11]:1200–1228).
“If you don’t have any of these genetic categories, you’re left with a morphologic classification,” she said, and most important would be the blast count. “If you have more than five percent blasts or more than two percent in the peripheral blood, or more than 10 percent blasts and more than five percent in the peripheral blood, you’re in the increased blast category.” In the WHO classification, MDS with fibrosis is also a distinct morphologic category. If the bone marrow is severely hypocellular for age, it is hypoplastic MDS (≤ 25 percent cellularity, age adjusted).
The ICC does not recognize hypoplastic MDS. “Biologically there is overlap between hypoplastic MDS and aplastic anemia,” Dr. Loghavi said. They’re thought of as two different diseases and they are, “but there’s a biological spectrum,” and the evidence shows that some patients with hypoplastic MDS respond well to immunosuppressive therapies and not necessarily to hypomethylating agents. For therapeutic purposes, the WHO authors thought “they would pull this category out to give the patients a chance” for response on immunosuppressive therapies.
If the patient is not in any of these categories, and there is increased blasts, then it is MDS with increased blasts. Designation of morphologic dysplasia, whether unilineage or multilineage, is optional in the WHO classification.
The ICC is similar, she said. The major difference is that in the ICC classification the two morphologic categories—hypoplastic MDS and MDS with fibrosis—are not recognized as distinct categories.
One major difference in the way the ICC and WHO classify MDS and AML, Dr. Loghavi said, is that a 10 percent blast count in the ICC takes a case out of the classic MDS category and places it in MDS/AML. “So if you have between 10 and 20 percent blasts without an AML-defining genetic alteration, it’s MDS/AML.” The WHO says that for clinical trial enrollment and therapeutic purposes, MDS-IB2 (the equivalent of MDS/AML) can be treated as AML at the discretion of the treating physician if the physician thinks the patient will respond better to AML-type therapy.
Why does the allelic state of TP53 in MDS matter?
A fortuitous finding came about as part of the International Prognostic Scoring System risk stratification project, Dr. Loghavi said (Bernard E, et al. Nat Med. 2020;26[10]:1549–1556). In patients who had multi-hit TP53 mutations, leukemia-free survival and overall survival outcomes were particularly dismal. Bernard, et al., write, “Surprisingly, monoallelic TP53 patients did not differ from TP53 wild-type patients with regard to overall survival and AML progression.”
Given the biology of TP53, “this makes perfect sense because TP53 is a tumor suppressor gene,” Dr. Loghavi said. “Typically in any type of malignancy, when there is a dysfunction of a tumor suppressor gene and you have one conserved copy, you normally don’t have a catastrophic event because the normal copy compensates for the abnormal copy. But once you lose both copies of a tumor suppressor gene, you lose the function of that gene. That is what happens with TP53.”
There are three ways to get to biallelic or multi-hit status, Dr. Loghavi said, first explaining her view that the multi-hit terminology is better. “Although on the science side we’re very sophisticated, in clinical practice nobody is doing single-cell sequencing. We’re doing bulk NGS, so there’s no way for us to know this is in fact a biallelic hit even if you have two mutations, unless the mutations are so close to one another physically that we can see them in the same reads in the NGS results. But we’re inferring that if there are two mutations, they’re in two different alleles because we know the biology of TP53.”
If there is one mutation and the other copy is lost by way of deletion, it can be seen on a routine karyotype or by FISH for TP53. MD Anderson Cancer Center, probably like most academic practices, she said, does not routinely perform FISH for TP53 unless “we see it on a karyotype, if we see an abnormality on 17p.” If there is a TP53 mutation at a high VAF or a complex karyotype with a TP53 mutation, “we will add a TP53 FISH to make sure we’re not missing a TP53 deletion.”
The most challenging scenario is having a copy neutral loss of heterozygosity of TP53 with a mutation, she said. The mutation is seen on the sequencing. But in most clinical laboratories, the assays don’t have the ability to detect copy neutral loss of heterozygosity. “This is the same kind of mechanism as a uniparental disomy. You have one mutant copy, and then the normal copy is deleted but the mutant copy is duplicated. So if you FISH for TP53 you’re going to see a normal signal because you have two copies of TP53 but both are abnormal.”
“If you have one mutation with a variant allele frequency of more than 50 percent,” she continued, “you can assume that the wild-type copy is deleted. You’re inferring and using it as a surrogate, but it normally works pretty well” (El Hussein S, et al. Cancers (Basel). 2022;14[22]:5690).
At MD Anderson, pathologists run a p53 immunohistochemistry on every newly diagnosed case of MDS or AML. “It’s much faster than sequencing and it’s a good screening tool,” Dr. Loghavi explained. A normal pattern of p53 staining is typically faint and patchy nuclear staining (Fig. 2, top left). “When you have a mutation—typically a missense or a splice-site mutation—the mutant copy is resistant to MDM2 degradation, so you get accumulation of p53 in the cell and the antibody picks up much higher levels of p53, and there is intense, dark nuclear staining [top right]” (Tashakori M, et al. Blood. 2022;140[1]:58–72).
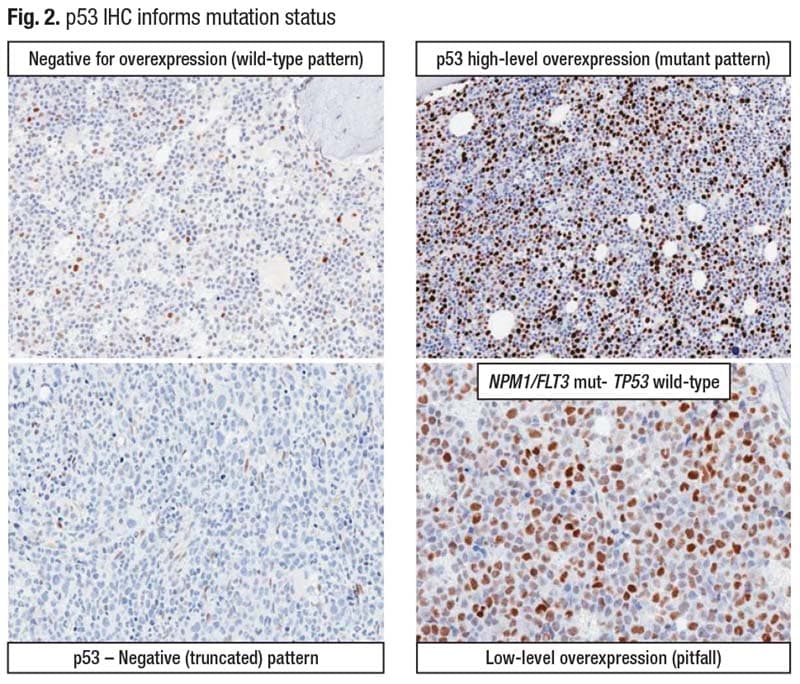
Staining intensity is important. “Moderate is not what we’re looking for,” Dr. Loghavi said. “It’s very bright, intense staining. And the number of cells that stain typically is commensurate with the variant allelic frequency of the mutation.” The higher the VAF, the more cells that stain intensely.
Another abnormal pattern—the null pattern—typically goes with a nonsense or early terminating frameshift mutation that results in the antibody not recognizing the protein (bottom left). There is weak staining in the spindle cells which are the endothelial cells, but the myeloid cells are negative.
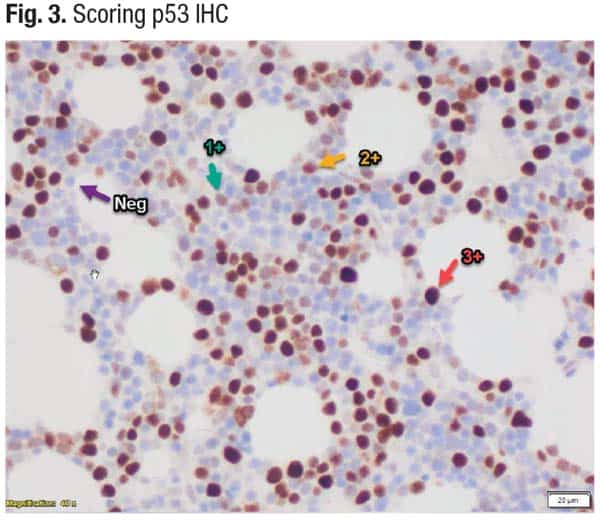
A p53 IHC stain is agnostic of the allelic status and thus won’t help in the classification of MDS. “You can say whether there’s a mutation or not, but you can’t tell if it’s biallelic or multi-hit,” she said.
In scoring a p53 IHC (Fig. 3), “the only intensity we care about is 3+ [red arrow],” Dr. Loghavi said. If more than 10 percent of cells are bright, intensely staining cells, there likely will be a mutation at a VAF of 10 percent or higher.
She returned to the case of the 51-year-old patient with increased blasts and fibrosis. Most important is whether there is a TP53 mutation, and there was a TP53 mutation at a variant allele frequency of 60 percent, she said, and a complex karyotype including del(17p). In the WHO classification, the case is MDS with biallelic TP53 inactivation (biallelic by definition because of the mutation and the 17p deletion). “Even though you have fibrosis, TP53 status trumps the presence of fibrosis,” Dr. Loghavi said.