
CAP TODAY and the Association for Molecular Pathology have teamed up to bring molecular case reports to CAP TODAY readers. AMP members write the reports using clinical cases from their own practices that show molecular testing’s important role in diagnosis, prognosis, and treatment. The following report comes from New York Institute of Technology, Memorial Sloan Kettering Cancer Center, Good Samaritan Hospital, and St. Francis Hospital and Heart Center—all in New York. If you would like to submit a case report, please send an email to the AMP at amp@amp.org. For more information about the AMP and all previously published case reports, visit www.amp.org.
Rachel Radigan, MPH; Alexa Finkelstein, BS
Tejus A. Bale, MD, PhD; Michael Zaidinski, BS
Marc K. Rosenblum, MD; Thomas Cawte, MD
Dong Zhang, PhD; Maria Plummer, MD
Wallace Chan, MD
February 2025—Intracranial mesenchymal tumors (IMT) are extremely rare and account for only 0.3 percent of soft tissue tumors.1 Most frequently, mesenchymal tumors occur in subcutaneous tissue; they are rarely described in the central nervous system. In addition, these tumors typically occur in young adults,1 with a median patient age of 14 (range: four to 70).2
Recently, it has become increasingly apparent that certain gene fusions may be shared among clinically unrelated sarcoma types. For example, the fusion genes EWSR1::CREB1 and EWSR1::ATF1 have been detected in angiomatoid fibrous histiocytoma, soft tissue and gastrointestinal clear cell sarcoma, primary pulmonary myxoid sarcoma, and hyalinizing clear cell carcinoma of the salivary gland.3 Intriguingly, EWSR1-rearrangement mesenchymal tumors in the central nervous system are rare and were found in only a small number of adolescents and young adults with intracranial myxoid mesenchymal tumors.4
Approximately one-third of soft tissue tumors are characterized by specific recurrent chromosomal translocations, leading to the generation of aberrant chimeric transcription factors. Most fusion transcripts define the associated sarcoma and thus can be used as a highly specific molecular diagnostic marker under the right clinical and pathological context.5
IMT with a FET::CREB fusion gene can mimic other intracranial tumors radiologically and histologically. The extra-axial location as well as the presence of a dural tail can result in the impression of a meningioma or solitary fibrous tumor/hemangiopericytoma. They can also resemble a meningioma microscopically as well owing to the presence of syncytial growth, meningioma-like whorls, rhabdoid cytology, or the presence of cords of epithelioid cells in a mucin-rich stroma. The expression of desmin and the presence of a FET::CREB fusion gene are useful in distinguishing IMT from meningioma immunohistochemically2 and molecularly.
Prior reports have demonstrated variable histopathological features including spindle cells in mucin or collagenous stroma, or epithelioid cells in a mucin-poor collagenous stroma. The immunohistochemical profile often demonstrates epithelial membrane antigen (EMA) and desmin positivity. One essential diagnostic feature defined by the World Health Organization classification is the intracranial location.6
The EWSR1::CREM fusion gene is a relatively novel variant of the EWSR1::CREB family of fusion genes, which was first identified in intracranial myxoid mesenchymal tumors (IMMT) in 2017 by Kao, et al.3 This type of tumor present at the interhemispheric frontal region is a rare feature that has been reported in only three cases, all of which have the EWSR1::CREM fusion gene.7 Here, we report a case of a 67-year-old female with the radiological presentation of a typical meningioma located in the anterior right frontal lobe. She was ultimately diagnosed with a myxoid spindle cell neoplasm that had characteristics of an IMT with the FET::CREB fusion gene.
Case presentation. A 67-year-old female presented to the emergency department in August 2023 after a witnessed seizure characterized by tonic-clonic movements and unresponsiveness for two to three minutes. She then experienced a brief respite followed by a second seizure. The patient was postictal on arrival to the emergency department but hemodynamically stable.
The patient underwent radiological assessment first with head CT scans without contrast, followed by MRI scans with and without contrast. The axial noncontrast head CT (Fig. 1A) demonstrated a homogeneous mildly hypodense 1.9-cm mass, resulting in mild impression upon the right frontal lobe with associated mild vasogenic edema. An associated small dural tail, which is similar to a meningioma, was denoted on an axial post-contrast T1-weighted brain MRI (Fig. 1B). This was also noted on sagittal post-contrast T1-weighted brain MRI (Fig. 1C).
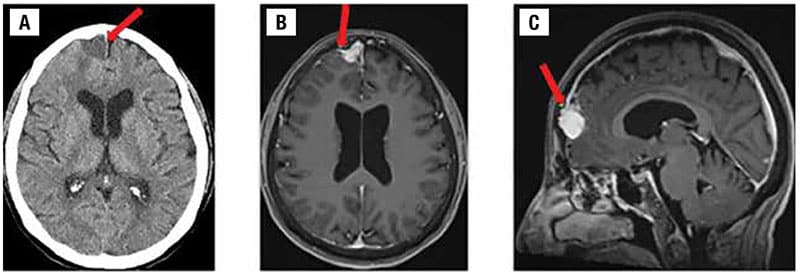
The patient subsequently underwent a right frontal craniotomy and brain tumor resection two days after the initial presentation. An intraoperative consultation grossly revealed a pale pink, irregularly shaped soft tissue mass measuring 1.3 × 0.3 cm. Microscopically, lesional tissue was reported as present. The excisional tissue was sent to Memorial Sloan Kettering Cancer Center for further workup. The tumor was described as a pale pink, rubbery nodule measuring 1.6 × 1.3 × 1.2 cm; one surface was slightly smooth, and the opposite surface was rough and irregular. The cut surface had a homogeneously pale pink appearance.
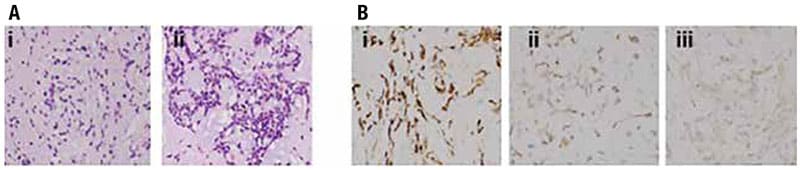
MSKCC analyzed the specimen microscopically and performed special staining and genetic testing via Archer analysis. Hematoxylin and eosin staining and light microscopy examination exhibited similar histopathology to tumors of this variant of IMT, which included solid and epithelioid features. More commonly, these tumors have been described as composed of spindle cell elements, in reticulated arrangements within a conspicuously myxoid matrix.8 The H&E staining showed a low-grade neoplasm with many spindle cells in a reticulated pattern present within a prominent myxoid background (Fig. 2A). Histopathology was reported to be consistent with a myxoid spindle cell neoplasm. Immunohistochemistry of the tumor showed the positive expression of desmin (Fig. 2B-i), EMA (Fig. 2B-ii), and CD99 (Fig. 2B-iii). The remaining immunohistochemical profile was negative for PR, SSTR2, SMA, STAT6, and MUC4 (data not shown). Ki-67, a proliferation marker for cancer cell division, was five percent (data not shown).
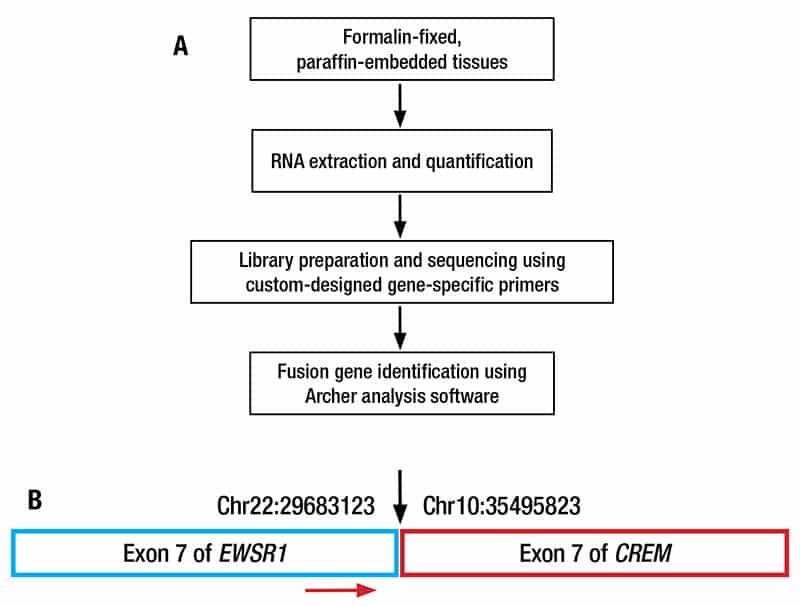
MSKCC also performed the MSK-Fusion assay to determine whether there is a known fusion gene in the patient’s tumor, which may provide a more definitive diagnosis (Fig. 3A).8 Briefly, RNA was extracted from formalin-fixed, paraffin-embedded tumor tissues and quantified. cDNA library was prepared from the extracted RNA using the Anchored Multiplex PCR technology and custom-designed gene-specific primers and sequenced. Fusion genes were identified using the Archer analysis software. Excitingly, this genetic workup demonstrated the presence of the EWSR1::CREM fusion gene (Fig. 3B).
Collectively, the final pathology report designated this as a myxoid spindle cell neoplasm characteristic of intracranial mesenchymal tumor with the fusion gene FET::CREB.
Postoperative imaging demonstrated a small resection cavity in the anterior right frontal region with reactive dural enhancement (data not shown). In December 2023, the patient had follow-up imaging for evaluation of the anterior frontal resection site. This revealed the presence of underlying encephalomalacia/gliosis. It was also noted that there was near resolution of fluid underlying the craniotomy with no new mass-like enhancement. Parenchymal volume loss and mild microvascular ischemic changes were also reported. The CT scan of the chest, abdomen, and pelvis showed no evidence of metastatic disease (data not shown). The patient’s 10-month postoperative surveillance MRI did not show any evidence of recurrence (data not shown). The patient is on antiseizure medication (levetiracetam) and has been neurologically stable without any further seizure episodes. She continues to follow up with a neurologist. The patient is doing well per her provider and will continue image surveillance.
Discussion. A few cases of intracranial mesenchymal tumors have been reported and have been variously termed intracranial angiomatoid fibrous histiocytoma or intracranial myxoid mesenchymal tumor.2 Prior case reports discussing intracranial mesenchymal tumors have disclosed both similarities with and differences from this case report. In 2017, Kao, et al., first described the novel EWSR1::CREM fusion gene, a variant of the EWSR1::CREB family of gene fusions.3
Most intracranial mesenchymal tumors have been reported in children and young adults in the second and third decades of life, with a median age of 17.2 Intriguingly, our case report is of a 67-year-old woman. Overall, females were affected more frequently than males, with a male-to-female ratio of 1:1.8 in both pediatric and adult populations.10 Prior tumors have been described as uniformly extra-axial or intraventricular with locations at the cerebral convexities, falx, lateral ventricles, tentorium, cerebellopontine angle, and spinal cord.2 The location of our patient’s neoplasm was designated as extra-axial and parafalcine, demonstrating similarities to prior cases.
Radiological reports have shown an extra-parenchymal lobulated mass without a dural tail,6 choroid plexus (glomus choroideum) meningioma,9 and contrast-enhancing, surrounding vasogenic edema, with no definitive dural attachment.11 The radiologic report of our patient’s tumor similarly showed vasogenic edema within the right frontal lobe; however, in contrast, a dural tail anteromedial to the right frontal lobe was reported, which gave the impression of a meningioma at first.
Histopathological analysis has varied among case reports and has revealed epithelioid morphology with mucin-rich stroma6 mostly showing “myxoid and/or fibromyxoid background stroma, with a variety of tumor architectural patterns, including cords and sheets, and diverse cell morphology, including epithelioid, rhabdoid, and stellate/spindle cell.” Additionally, histopathology reports have shown collagenous or myxoid stroma; internodular septae; lymphoplasmacytic cuffing; and hemangioma-like, or staghorn/hemangiopericytoma-like, vasculature.2,9 Our patient’s tumor demonstrated myxoid background stroma with spindle cells. Immunohistochemistry studies of prior cases have also shown positive expression of EMA and desmin,6 EMA and CD99,9 or desmin, EMA, CD99, MUC4, and synaptophysin.2 Similarly, our patient’s tumor showed the expression of EMA, CD99, and desmin (Fig. 2B).
In terms of genetic fusions, most intracranial mesenchymal tumors have shown the presence of a chimeric fusion of a FET family gene, usually EWSR1, to a CREB family transcription factor of ATF1, CREB1, or CREM.2,6,12 A report of an EWSR1::CREM fusion demonstrated an epigenetic DNA-methylation profile.6 Based on the genetic profiling done on the tumor tissue of our case, the tumor was positive for EWSR1::CREM fusion (Fig. 3).
Studies have shown that a chromosomal translocation producing the EWSR1::CREM fusion gene and a chimeric protein has resulted in divergent malignancies including both low-grade to highly malignant cancers. The molecular mechanisms related to the chimeric protein’s oncogenic properties were studied using siRNA-mediated depletion of the chimeric EWSR1-CREM protein and showed altered expression of 712 genes. Many of these genes were found to be involved in cell cycle, proliferation, and migration.13
Ewing sarcoma breakpoint region 1 (EWSR1) gene is located on chromosome 22q12 and contains 17 exons, which encodes a highly conserved protein with 656 amino acid. EWSR1 protein belongs to the FET family of proteins, which also includes FUS and TAF15. This family of proteins is a highly conserved group of multifunctional, RNA-binding proteins that play a role in regulating transcription, RNA processing, mRNA cytoplasmic destination, DNA repair mechanisms, meiotic and mitotic cell division, and cellular aging. They have been shown to promote various sarcomas if chromosomally rearranged.14,15
Cyclic adenosine monophosphate (cAMP) responsive element modulator (CREM) is a gene located on chromosome 10p11.21.2 CREM encodes a protein that is part of the CREB family of transcription factors, including ATF1 and CREB.9 CREM binds to the cAMP responsive elements. Through alternative promoter and translation initiation sites, CREM can exert spatial and temporal specificity to cAMP responsiveness. CREM gene has been found to produce various protein isoforms through alternative splicing, which can act as transcription activators or repressors.16
 The long-term outcome of IMT patients with the FET::CREB family of gene fusions has varied. Local recurrence was common, ranging from six months to 120 months,7 with an average of 12 months, and a median overall survival greater than 60 months.2 This patient had a follow-up imaging four months after resection; no recurrence was noted at that time.
The long-term outcome of IMT patients with the FET::CREB family of gene fusions has varied. Local recurrence was common, ranging from six months to 120 months,7 with an average of 12 months, and a median overall survival greater than 60 months.2 This patient had a follow-up imaging four months after resection; no recurrence was noted at that time.
In terms of treatment, of the 44 cases that have been reported, 34 individuals underwent a gross total resection. Additionally, seven individuals underwent certain types of adjuvant therapy including chemotherapy, radiation therapy, or both.10
A previous study by Kaprio, et al., using siRNA-mediated depletion of EWSR1-CREM demonstrated the importance of the loss of ornithine decarboxylase 1 (ODC1), which is critically involved in polyamine synthesis.13 Polyamine biosynthesis is a rate-limiting step that regulates cell proliferation. The results from this study confirmed that the oncogenic properties of EWSR1-CREM are likely mediated by ODC1. These findings provide novel insights into the pathogenesis of tumors harboring an EWSR1::CREM fusion gene, hopefully facilitating the development of novel therapeutic strategies. For example, allicin, a reactive sulfur species from garlic (Allium sativum L.), is hydrophobic in nature and can efficiently cross cellular membranes. Schultz, et al., reported that allicin targeted ODC1 in neuroblastoma cells.17 Remarkably, allicin inhibited the enzyme activity with a 23,000-fold higher potency than difluoromethylornithine.18 This demonstrates a potential therapeutic use for allicin in treating IMT.
In summary, we report a novel intracranial mesenchymal tumor with the EWSR1::CREM fusion gene that has similarities to and differences from prior case reports of this tumor type. This case is even more unique based on the older age of our patient. IMT is a rare entity that may present with diagnostic challenges owing to its rarity and its close resemblance to other tumors. While this report has the potential benefit to aid in future diagnoses of similar cases, continued research is still needed.
Abbreviations: EWSR1: Ewing Sarcoma Breakpoint Region 1 Gene; CREM: cAMP Responsive Element Modulator; CREB: cAMP Response Element Binding Protein; ATF1: Activating Transcription Factor 1; FET: FUS/EWS/Taf15 fusion oncoproteins; FLAIR: Fluid Attenuated Inversion Recovery; CD99: Cluster of Differentiation 99; PR: Progesterone Receptor; SSTR2: Somatostatin Receptor 2; SMA: Smooth Muscle Actin; STAT6: Signal Transducer and Activator of Transcription 6; MUC4: Mucin 4.
- D’Antonio F, Rossi S, Giovannoni I, et al. Case report: Remarkable breakthrough: successful treatment of a rare intracranial mesenchymal, FET::CREB fusion-positive tumor treated with patient-tailored multimodal therapy. Front Oncol. 2023;13:1203994.
- Sloan EA, Chiang J, Villanueva-Meyer JE, et al. Intracranial mesenchymal tumor with FET-CREB fusion—a unifying diagnosis for the spectrum of intracranial myxoid mesenchymal tumors and angiomatoid fibrous histiocytoma-like neoplasms. Brain Pathol. 2021;31(4):e12918.
- Kao YC, Sung YS, Zhang L, et al. EWSR1 fusions with CREB family transcription factors define a novel myxoid mesenchymal tumor with predilection for intracranial location. Am J Surg Pathol. 2017;41(4):482–490.
- Adams JW, Malicki D, Levy M, Crawford JR. Rare intracranial EWSR1-rearranged myxoid mesenchymal tumour in a teenager. BMJ Case Rep. 2021;14(8):e245282.
- Cerrone M, Cantile M, Collina F, et al. Molecular strategies for detecting chromosomal translocations in soft tissue tumors (review). Int J Mol Med. 2014;33(6):1379–1391.
- Tauziède-Espariat A, Pierron G, Guillemot D, et al. An extracranial CNS presentation of the emerging “intracranial” mesenchymal tumor, FET:CREB-fusion positive. Brain Tumor Pathol. 2023;40(1):35–39.
- Domingo RA, Vivas-Buitrago T, Jentoft M, Quinones-Hinojosa A. Intracranial myxoid mesenchymal tumor/myxoid subtype angiomatous fibrous histiocytoma: diagnostic and prognostic challenges. Neurosurgery. 2021;88(1):E114–E122.
- Benayed R, Offin M, Mullaney K, et al. High yield of RNA sequencing for targetable kinase fusions in lung adenocarcinomas with no mitogenic driver alteration detected by DNA sequencing and low tumor mutation burden. Clin Cancer Res. 2019;25(15):4712–4722.
- Ozkizilkaya HI, Johnson JM, O’Brien BJ, et al. Intracranial mesenchymal tumor, FET::CREB fusion-positive in the lateral ventricle. Neurooncol Adv. 2023;5(1):vdad026.
- Sasaki M, Hirono S, Gao Y, et al. Clinicopathological and genomic features of pediatric intracranial myxoid mesenchymal tumor with both of EWSR1-CREM gene fusion and MAP3K13 mutation: a case report and comparison with adult cases in the literature. NMC Case Rep J. 2022;9:101–109.
- Bale TA, Oviedo A, Kozakewich H, et al. Intracranial myxoid mesenchymal tumors with EWSR1–CREB family gene fusions: myxoid variant of angiomatoid fibrous histiocytoma or novel entity? Brain Pathol. 2018;28(2):183–191.
- Tauziède-Espariat A, Hasty L, Métais A, Varlet P. Mesenchymal non-meningothelial tumors of the central nervous system: a literature review and diagnostic update of novelties and emerging entities. Acta Neuropathol Commun. 2023;11(1):22.
- Kaprio H, Siddiqui A, Saustila L, Heuser VD, Gardberg M. The oncogenic properties of the EWSR1::CREM fusion gene are associated with polyamine metabolism. Sci Rep. 2023;13(1):4884.
- Schwartz JC, Cech TR, Parker RR. Biochemical properties and biological functions of FET proteins. Annu Rev Biochem. 2015;84:355–379.
- Jo VY. EWSR1 fusions: Ewing sarcoma and beyond. Cancer Cytopathol. 2020;128(4):229–231.
- Kaprio H, Heuser VD, Orte K, Tukiainen M, Leivo I, Gardberg M. Expression of transcription factor CREM in human tissues. J Histochem Cytochem. 2021;69(8):495–509.
- Schultz CR, Gruhlke MCH, Slusarenko AJ, Bachmann AS. Allicin, a potent new ornithine decarboxylase inhibitor in neuroblastoma cells. J Nat Prod. 2020;83(8):2518–2527.
- Kong X, Gong S, Su L, Li C, Kong Y. Neuroprotective effects of allicin on ischemia-reperfusion brain injury. Oncotarget. 2017;8(61):104492–104507.
Rachel Radigan and Alexa Finkelstein (equally contributing authors) are in the Department of Biomedical Sciences, College of Osteopathic Medicine, New York Institute of Technology, Old Westbury, NY. Dr. Bale, Michael Zaidinski, and Dr. Rosenblum are in the Department of Pathology and Laboratory Medicine (neuropathology and diagnostic molecular pathology), Memorial Sloan Kettering Cancer Center, New York. Dr. Cawte is in the Department of Pathology, Good Samaritan University Hospital, West Islip, NY. Dr. Zhang is in the Department of Biomedical Sciences and Dr. Plummer is in the Department of Clinical Specialties, College of Osteopathic Medicine, and the Center for Cancer Research, New York Institute of Technology, Old Westbury, NY. Dr. Chan is with the Cancer Institute of St. Francis Hospital and Heart Center, East Hills, NY.