
![]() CAP TODAY and the Association for Molecular Pathology have teamed up to bring molecular case reports to CAP TODAY readers. AMP members write the reports using clinical cases from their own practices that show molecular testing’s important role in diagnosis, prognosis, and treatment. The following report comes from Akron (Ohio) Children’s Hospital and Northeast Ohio Medical University. If you would like to submit a case report, please send an email to the AMP at amp@amp.org. For more information about the AMP and all previously published case reports, visit www.amp.org.
CAP TODAY and the Association for Molecular Pathology have teamed up to bring molecular case reports to CAP TODAY readers. AMP members write the reports using clinical cases from their own practices that show molecular testing’s important role in diagnosis, prognosis, and treatment. The following report comes from Akron (Ohio) Children’s Hospital and Northeast Ohio Medical University. If you would like to submit a case report, please send an email to the AMP at amp@amp.org. For more information about the AMP and all previously published case reports, visit www.amp.org.
Jie Xu, PhD
Ilka Warshawsky, MD, PhD
Sam Dougaparsad, PhD
Catherine Melver, MD
Carrie Costin, MD
March 2020—Prader-Willi syndrome (PWS) is a rare genetic multisystem disorder with a reported incidence of approximately one in 15,000. It is characterized by severe infantile hypotonia with feeding problems, global developmental delay and mental deficiency, behavior problems, small hands and feet, hypogonadism and hyperphagia leading to marked obesity in early childhood, and a characteristic face. PWS is caused by one of several genetic mechanisms that results in loss of expression of imprinted genes in the paternally derived PWS/Angelman syndrome region at chromosome 15q11.2q13. Currently there is no national guideline or standard of practice for a PWS genetic testing algorithm.1
Genetic diagnosis of PWS commonly begins with methylation-specific PCR (MSPCR) of the SNRPN locus. MSPCR detects more than 99 percent of PWS mutations caused by a paternal deletion at chromosome 15q11.2q13 (about 60 percent of cases), chromosome 15 maternal uniparental disomy (mUPD 15) (about 36 percent of cases), and an imprinting center defect (about four percent of cases).2,3 MSPCR, however, does not differentiate the three genetic subtypes, which is important in genetic counseling for recurrence risks to sibs. Recurrence risk is less than one percent if the affected child has a paternal deletion or mUPD 15 and up to 50 percent if the affected child has an imprinting center defect.
There are two types of typical 15q11.2q13 deletions: type one deletions are approximately 6.6 Mb in size and involve proximal BP1 and distal BP3 breakpoints; type two deletions are about 5.3 Mb in size and involve proximal BP2 and distal BP3 breakpoints. Atypical deletions occur in seven to nine percent of those with deletions and are smaller or larger than type one and type two deletions.3 mUPD 15 can have three subtypes: heterodisomy, total isodisomy, or segmental isodisomy.3
Defining the mechanism of MSPCR-positive PWS frequently starts with FISH to look for a chromosome 15q deletion. If FISH is negative, microsatellite analysis is often performed next to look for mUPD 15; it compares tracts of repetitive DNA along chromosome 15 from both parents and the child. If microsatellite analysis is negative, DNA sequencing is performed to look for an imprinting center defect. A newer cytogenomics method called single nucleotide polymorphism (SNP) chromosomal microarray can detect genomic copy number changes (i.e. deletions or duplications), uniparental disomy, and region(s) of homozygosity.
Here we discuss two cases and suggest that SNP chromosomal microarray as a follow-up to MSPCR-positive PWS allows for a more accurate diagnosis of the three genetic subtypes of PWS. Additionally, trio (father, mother, proband) SNP chromosomal microarray can define the three subtypes of mUPD 15 by using a combination of SNP genotypes and Mendelian Inheritance Error (MIE) analysis. MIE analysis is derived from a statistical algorithm developed by Marshall, et al., at the University of Edinburgh.4 This analysis tool is available using the SNP chromosomal microarray CytoScan Dx assay (Thermo Fisher Scientific) and scores parent-offspring relationships based on SNP genotyping markers. A confidence value for Mendelian consistencies is computed for each autosome. In general, normal MIE score values should be less than one percent for each autosome (https://assets.thermofisher.com/TFS-Assets/LSG/manuals/ChAS_Manual.pdf; pp. 97–100). We propose that positive MSPCR followed by SNP chromosomal microarray may improve genotype-phenotype correlations and allows for surveillance of a second genetic defect in cases with total or segmental isodisomy PWS.
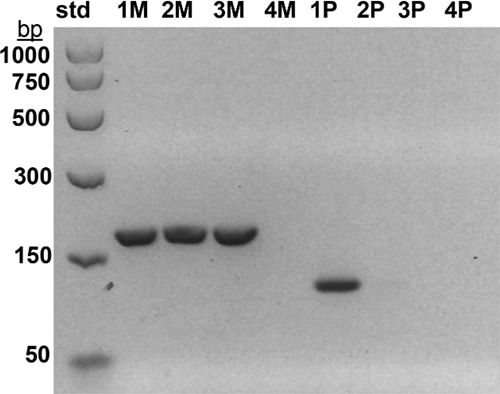
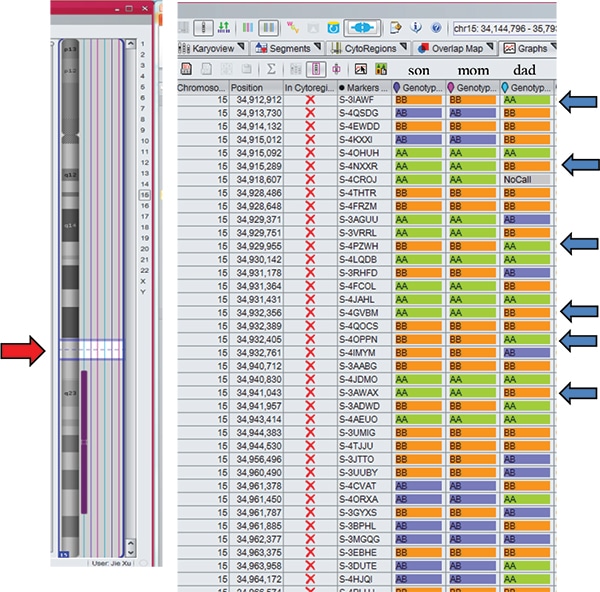
Case studies. Case 1: A two-week-old male born to a 44-year-old woman presented with hypotonia, cryptorchidism, poor feeding, and dysmorphic features, and was small for gestational age. MSPCR amplified only a 174-bp fragment from the maternal methylated SNRPN gene, consistent with PWS (Fig. 1). SNP chromosomal microarray analysis was performed using the CytoScan Dx assay and did not show deletion of 15q11.2q13 or any clinically relevant copy number changes. However, an approximately 28-Mb region of homozygosity was seen in interstitial 15q: 15q22.31q26.1. Trio MIE analysis showed chromosome 15 MIEs were high for the trio (7.45 percent) and father (7.28 percent) and low for the mother (0.01 percent), indicative of mUPD 15.4 A graphical representation of genotypes for the trio is displayed in Fig. 2 (as a visual confirmation of the MIE value). The proband and mother shared identical genotype calls for proximal 15q: 15q11.2q22.31 and distal 15q: 15q26.1q26.3, consistent with maternal heterodisomy (Fig. 2a). The proband had homozygous genotype calls for interstitial 15q: 15q22.31q26.1, consistent with maternal segmental isodisomy (Fig. 2b).
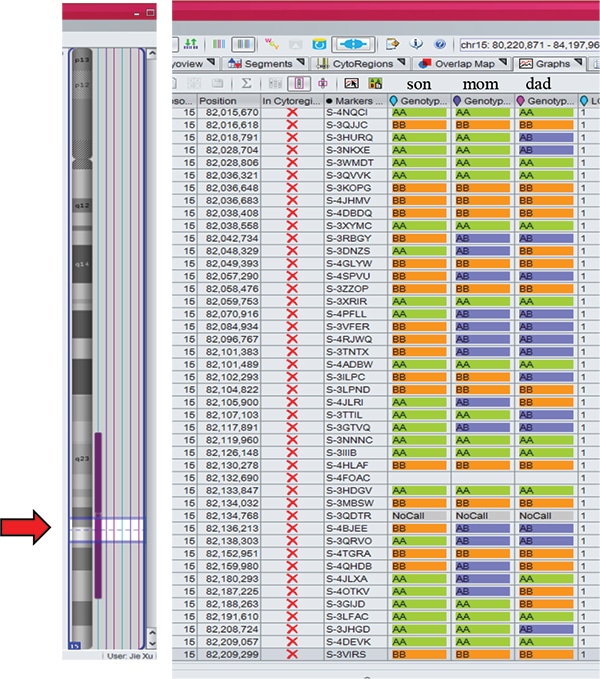
Case 2: A three-week-old male born to a 39-year-old woman presented with hypotonia, feeding difficulties, and undescended testes. MSPCR was consistent with PWS. FISH was negative for deletion of SNRPN at 15q11.2q13. The CytoScan Dx assay did not show deletion of 15q11.2q13 but did show an approximately 11.8-Mb region of homozygosity in distal 15q: 15q26.1q26.3. Additionally, an approximately 136-kb duplication of 2q24.2 involving three OMIM genes (CD302, MARCH7, and LY75) was seen. Trio SNP chromosomal microarray studies showed the duplication was maternally inherited and of uncertain significance. Trio MIE analysis showed chromosome 15 MIEs were high for the trio (6.26 percent) and father (5.94 percent) and low for the mother (0.01 percent), indicating mUPD 15. The proband and mother shared identical genotype calls for proximal 15q: 15q11.2q26.1, confirming maternal heterodisomy. The proband had homozygous genotype calls for distal 15q: 15q26.1q26.3, consistent with maternal segmental isodisomy.
In summary, both PWS cases were due to mUPD 15 with segmental isodisomy (i.e. regions of both maternal heterodisomy and maternal isodisomy) and assisted in genetic counseling of an approximately one percent recurrence risk. The finding of segmental isodisomy may impact medical surveillance for a second genetic condition if the mother is a carrier of a recessive disorder allele in the region of homozygosity, leading to two copies of the recessive allele. Advanced maternal age, seen in both cases, has been associated with mUPD 15.3 Because advanced maternal age is becoming more prevalent in Western societies, this may have an impact on the frequency of PWS molecular classes. Those with mUPD 15 have been reported to have higher verbal IQs and may be more prone to psychosis and autism.3
Discussion. What testing algorithm to use as an initial follow-up for MSPCR-positive PWS cases depends on many factors, including the clinical indication, family history, availability of parental samples, degree of clinical urgency, available testing platforms, laboratory testing volumes, and test costs. Advances in genetic technology have allowed for more accurate and reliable identification of the molecular genetic subtypes and improved genotype-phenotype correlations in PWS. In our setting, SNP chromosomal microarray analysis is preferable as a follow-up to MSPCR-positive PWS cases for the following six reasons:
First, unlike FISH for 15q11.2q13 deletions, SNP chromosomal microarray can detect and differentiate typical type one and type two and atypical 15q deletions which differ in clinical phenotypes. Adaptive behavior, obsessive-compulsive behaviors, and reading, math, and visual-motor integration assessments are worse in individuals with type one deletions than in individuals with type two deletions or mUPD 15. Four genes (TUBGCP5, CYFIP1, NIPA1, NIPA2) in the chromosome 15 region between BP1 and BP2 are deleted in individuals with type one deletions and are associated with autism, neurodevelopmental disturbances, and developmental delays.3
Second, trio SNP chromosomal microarray/MIE analyses can detect three subclasses of uniparental disomy (heterodisomy, total isodisomy, and segmental isodisomy) while microsatellite analysis may not if the microsatellite markers used are not of high enough density and do not cover the entire chromosome. More than 76 cases (including ours) of segmental isodisomy have been reported with 1-3 isodisomy segments ranging in size from about five to 67 Mb and covering six to 84 percent of chromosome 15.3,5-8 Segmental isodisomy can occur in any region of 15q. Similar to patients with total isodisomy, the identification of segmental isodisomy may place patients at a greater risk for a second genetic disorder if the mother carries a recessive mutant allele in the region of homozygosity.
Third, SNP chromosomal microarray can detect imprinting center defect microdeletions (about 25 percent of ICD cases).3
Fourth, SNP chromosomal microarray may provide faster turnaround times (about one week in our laboratory when needed). This may be significant, especially for cases that need parental studies for both mUPD 15 and copy number changes initially detected in the proband (as illustrated in case No. 2). Sequential FISH, microsatellite analysis, and sequencing tests may take longer. Although methylation-specific multiplex ligation-dependent probe amplification (MS-MLPA) has been reported to detect and differentiate type one and type two deletions as well as non-deletion status, it does not differentiate between mUPD 15 and imprinting defects.2,9 Additionally, for our laboratory, annual volumes of samples for PWS (and Angelman syndrome) do not justify implementation of MS-MLPA.
Fifth, SNP chromosomal microarray scans the whole genome (not just chromosome 15q). This may provide additional information for diagnosis or further investigation. For example, detection of copy number changes and/or region(s) of homozygosity on different chromosome(s) may lead to further investigations (e.g. sequencing) and identification of novel disease-causing genes and phenotypic expansion.10
Of note, SNP chromosomal microarray, as a follow-up test to MSPCR-positive PWS, is not intended to detect the “PWS-like phenotype”-associated cytogenomic rearrangements, such as an interstitial deletion of 6q16.2, 1p36 deletion, 16p11.2 deletion, duplication Xq27.2qter, and deletion 10q26, or mimics of parts of the PWS phenotype,2 unless these rearrangements are in addition to deletion 15q11.2q13.
The SNP chromosomal microarray analysis can lead to incidental findings. This possibility should be discussed during pretesting genetic counseling.
Finally, proband-only SNP chromosomal microarray analysis may detect abnormalities in 86 percent of PWS patients,2 while trio SNP chromosomal microarray analysis may increase the detection rate to 97 percent.
- Smith A, Hung D. The dilemma of diagnostic testing for Prader-Willi syndrome. Transl Pediatr. 2017;6(1):46–56.
- Driscoll DJ, Miller JL, Schwartz S, Cassidy SB. Prader-Willi syndrome. In: Adam MP, Ardinger HH, Pagon RA, et al., eds. GeneReviews. Seattle: University of Washington, Seattle; 1993–2019. https://www.ncbi.nlm.nih.gov/books/NBK1116/. Posted Oct. 6, 1998. Updated Dec. 14, 2017.
- Butler MG, Hartin SN, Hossain WA, et al. Molecular genetic classification in Prader-Willi syndrome: a multisite cohort study. J Med Genet. 2019;56(3):149–153.
- Marshall TC, Slate J, Kruuk LE, Pemberton JM. Statistical confidence for likelihood-based paternity inference in natural populations. Mol Ecol. 1998;7(5):639–655.
- Liu S, Zhang K, Song F, et al. Uniparental disomy of chromosome 15 in two cases by chromosome microarray: a lesson worth thinking. Cytogenet Genome Res. 2017;152(1):1–8.
- Papenhausen P, Schwartz S, Risheg H, et al. UPD detection using homozygosity profiling with a SNP genotyping microarray. Am J Med Genet A. 2011;155A(4):757–768.
- Wang JC, Ross L, Mahon LW, et al. Regions of homozygosity identified by oligonucleotide SNP arrays: evaluating the incidence and clinical utility. Eur J Hum Genet. 2015;23(5):663–671.
- Zeesman S, McCready E, Sadikovic B, Nowaczyk MJ. Prader-Willi syndrome and Tay-Sachs disease in association with mixed maternal uniparental isodisomy and heterodisomy 15 in a girl who also had isochromosome Xq. Am J Med Genet A. 2015;167A(1):180–184.
- Mahmoud R, Singh P, Weiss L, et al. Newborn screening for Prader-Willi syndrome is feasible: early diagnosis for better outcomes. Am J Med Genet. 2019;179(1):29–36.
- Alabdullatif MA, Al Dhaibani MA, Khassawneh MY, El-Hattab AW. Chromosomal microarray in a highly consanguineous population: diagnostic yield, utility of regions of homozygosity, and novel mutations. Clin Genet. 2017;91(4):616–622.
- Kubota T, Das S, Christian SL, Baylin SB, Herman JG, Ledbetter DH. Methylation-specific PCR simplifies imprinting analysis. Nat Genet. 1997;16(1):16–17.
Acknowledgments: We wish to thank Gail Dunphy, Julie Boles, Sue Arnold, and James Malone for outstanding assistance with this work.
Dr. Xu is a clinical cytogeneticist and director of cytogenetics, Akron (Ohio) Children’s Hospital, and professor of pathology, Northeast Ohio Medical University. Dr. Warshawsky is a molecular pathologist and director of molecular diagnostics, Akron Children’s Hospital, and assistant professor of pathology, Northeast Ohio Medical University. Dr. Dougaparsad is a reproductive health specialist, Thermo Fisher Scientific, Santa Clara, Calif. Dr. Melver and Dr. Costin are medical geneticists at Akron Children’s Hospital.
Test yourself
[tabs type=”horizontal”][tabs_head][tab_title]Here are three questions taken from the case report.[/tab_title][tab_title]Answers[/tab_title][/tabs_head][tab]
1. What is the first test commonly used for diagnosis of Prader-Willi syndrome?
a. FISH test for deletion 15q11.2q13.
b. Microarray testing for UPD 15.
c. Methylation-specific PCR of the SNRPN locus.
d. DNA sequencing for imprinting center defect.
2. What are cytogenomic mechanisms that can cause Prader-Willi syndrome?
a. Paternal deletion 15q11.2q13.
b. Maternal UPD 15.
c. Imprinting center defect.
d. All of the above.
3. What are potential advantages of SNP chromosomal microarray analysis over conventional FISH/DNA tests for methylation-specific PCR-positive PWS cases?
a. Microarray can detect and differentiate typical type one and type two and atypical 15q deletions; FISH cannot differentiate type one and two deletions and may miss atypical and small deletions.
b. Trio SNP genotyping/Mendelian Inheritance Error analyses can detect all types of uniparental disomy (heterodisomy, total isodisomy, segmental isodisomy) while microsatellite analysis may not.
c. Microarray may provide faster turnaround times for cases that need parental studies for both uniparental disomy and copy number changes initially detected in the proband.
d. Microarray scans the whole genome for clinically relevant rearrangements (not just 15q), which may provide additional diagnostic information.
e. Proband only SNP chromosomal microarray may detect abnormalities in 86 percent of PWS patients, whereas trio analyses may increase the detection rate to 97 percent.
f. All of the above.
[/tab]
[tab]
1. What is the first test commonly used for diagnosis of Prader-Willi syndrome?
a. FISH test for deletion 15q11.2q13.
b. Microarray testing for UPD 15.
c. Methylation-specific PCR of the SNRPN locus.
d. DNA sequencing for imprinting center defect.
2. What are cytogenomic mechanisms that can cause Prader-Willi syndrome?
a. Paternal deletion 15q11.2q13.
b. Maternal UPD 15.
c. Imprinting center defect.
d. All of the above.
3. What are potential advantages of SNP chromosomal microarray analysis over conventional FISH/DNA tests for methylation-specific PCR-positive PWS cases?
a. Microarray can detect and differentiate typical type one and type two and atypical 15q deletions; FISH cannot differentiate type one and two deletions and may miss atypical and small deletions.
b. Trio SNP genotyping/Mendelian Inheritance Error analyses can detect all types of uniparental disomy (heterodisomy, total isodisomy, segmental isodisomy) while microsatellite analysis may not.
c. Microarray may provide faster turnaround times for cases that need parental studies for both uniparental disomy and copy number changes initially detected in the proband.
d. Microarray scans the whole genome for clinically relevant rearrangements (not just 15q), which may provide additional diagnostic information.
e. Proband only SNP chromosomal microarray may detect abnormalities in 86 percent of PWS patients, whereas trio analyses may increase the detection rate to 97 percent.
f. All of the above.
[/tab]
[/tabs]