
CAP TODAY and the Association for Molecular Pathology have teamed up to bring molecular case reports to CAP TODAY readers. AMP members write the reports using clinical cases from their own practices that show molecular testing’s important role in diagnosis, prognosis, and treatment. The following report comes from the Department of Pathology and Laboratory Medicine, Beaumont Health, Royal Oak, Mich. If you would like to submit a case report, please send an email to the AMP at amp@amp.org. For more information about the AMP and all previously published case reports, visit www.amp.org.
Kristle Haberichter, DO; Ann Marie Blenc, MD; Anne Prada, MB(ASCP); Bobby L. Boyanton Jr., MD
August 2019–Newly discovered pathogenic variants in BCR-ABL1-negative myeloproliferative neoplasms (i.e. polycythemia vera, essential thrombocythemia, primary myelofibrosis) led to recent revisions of the World Health Organization diagnostic criteria.1,2 Initially JAK2 V617F, MPL (MPL W515K/L), and calreticulin (CALR) exon 9 gene pathogenic variants were deemed mutually exclusive in patients with essential thrombocythemia and primary myelofibrosis. However, coexisting somatic variants in both JAK2 V617F and CALR have been reported with variable frequency, ranging from less than one percent and up to 6.8 percent depending on the employed molecular technique.3,4 Several somatic CALR variants have been noted to occur with JAK2 V617F. However, there are few reports in the current literature describing a coexisting JAK2 V617F pathogenic variant along with a germline CALR nonpathogenic variant. Herein, we present a patient with newly diagnosed essential thrombocythemia with a low-allele fraction somatic JAK2 V617F pathogenic variant and a likely germline CALR nonpathogenic variant.![]()
Case. A 67-year-old female presented with a nine-year history of persistent thrombocytosis (platelet counts ranged from 450,000 to 500,000 per microliter) without evidence of thrombosis or bleeding. JAK2 V617F, JAK2 exon 12, and CALR variant analyses were performed on peripheral blood. JAK2 exon 12 variant analysis was negative. A low-allele fraction (approximately one percent) pathogenic variant involving JAK2 V617F was detected (Fig. 1). CALR exon 9 variant analysis was positive for a 9-basepair deletion (47 percent allele fraction, likely germline heterozygous) (Fig. 2). Repeat testing by a national reference laboratory using polymerase chain reaction and fragment analysis confirmed the 9-bp deletion and noted the finding to likely represent a previously described in-frame germline polymorphism of no known clinical significance. Bidirectional Sanger sequencing using an ABI 310 genetic analyzer disclosed a novel 9-bp deletion (CALR: c.1280_1288del) predicted to be of no clinical significance due to preservation of the C-terminal KDEL (endoplasmic reticulum retention sequence) signal sequence. A bone marrow biopsy revealed a mildly hypercellular marrow (about 50 percent cellularity) with megakaryocytic hyperplasia. Occasional megakaryocyte clusters and frequent large, atypical forms with abundant mature cytoplasm and hypersegmented nuclei were identified (Fig. 3, page 28). The granulocytic and erythroid lineages showed no morphologic abnormalities. Cytogenetic studies demonstrated a normal female karyotype. The bone marrow findings in conjunction with a known JAK2 V617F pathogenic variant are diagnostic of a myeloproliferative neoplasm, most consistent with essential thrombocythemia.
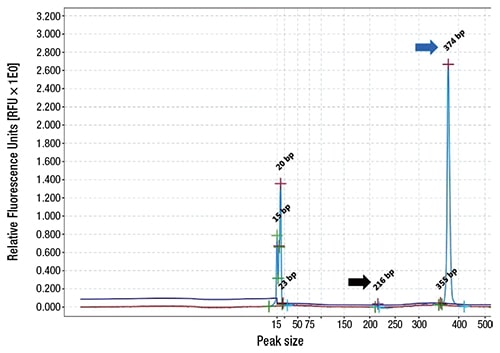
Discussion. Two landmark studies simultaneously identified the presence of recurrent somatic CALR pathogenic variants in the majority of JAK2 V617F-negative and MPL-negative essential thrombocythemia patients.5,6 The CALR gene (chromosome 19p13.2) codes for a protein involved with calcium homeostasis and glycoprotein folding within the endoplasmic reticulum.3,6 Somatic CALR pathogenic variants (insertions/deletions in exon 9) cause a frameshift in the coding sequence, resulting in loss of the endoplasmic reticulum retention sequence and the development of a novel C-terminus sequence.5-7 This novel C-terminal sequence leads to gain-of-function and activation of the thrombopoietin receptor with downstream cellular signaling via the JAK-STAT pathway.7 Numerous somatic CALR frameshift pathogenic variants have been described; the two most frequent (about 85 percent of cases) are the result of either a 52-basepair deletion (type 1) or a 5-basepair insertion (type 2).5 In contrast, in-frame germline CALR variants occur in 3-basepair multiples, allowing for preservation of the reading frame and retention of normal KDEL C-terminal sequence.5 The frequency of germline CALR variants has been documented to occur in less than one percent of patients.5,8
A recent study by Szuber, et al., investigated the identification, confirmation, and significance of germline CALR variants.8 They examined 2,000 peripheral blood samples and identified 12 samples (0.6 percent) that demonstrated unique variants, representing either a 12-basepair deletion (n=10) or a 9-basepair deletion (n=2). In those patients for whom confirmatory testing was performed on buccal cell samples, they were able to show 100 percent congruity (n=7) at the same allele fraction of about 50 percent, therefore confirming the germline nature of the variants. Within the same patient cohort a few patients had coexisting JAK2 V617F (n=3) or somatic CALR variants (n=1) and a clinical diagnosis of a myeloproliferative neoplasm. None of the patients with an isolated germline CALR variant had a myeloproliferative neoplasm at the time of diagnostic testing, signifying that such variants were likely benign.
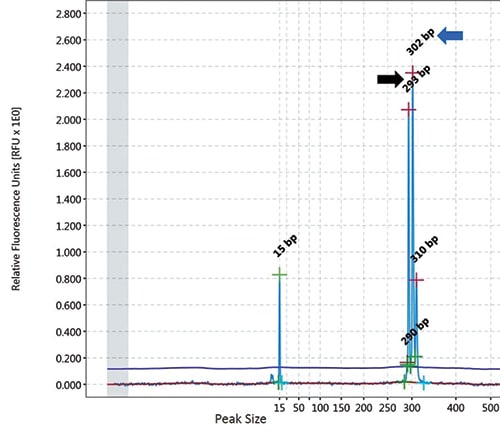
The reported coexisting positivity rate likely does not reflect the true positivity rate, as not all patients with JAK2 V617F variants are tested for CALR variants, especially given the original assumption of mutual exclusivity and the use of predefined testing algorithms. Furthermore, testing for all possible variants in patients with suspected myeloproliferative neoplasms is not suggested because the low positivity rate does not justify the cost of testing.
It has been noted that patients with somatic CALR pathogenic variants have a more indolent clinical course with a lower thrombosis risk and longer overall survival in comparison to those patients with JAK2 V617F pathogenic variants.5,6,12 However, the exact clinical significance of coexisting variants has yet to be clearly defined secondary to the limited number of reported cases.
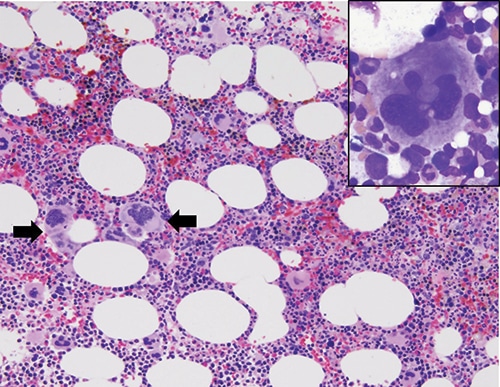
Overall, this case study represents the importance of molecular testing in the diagnosis of myeloproliferative neoplasms. It is imperative to properly characterize the type of CALR variant detected. Our case highlights this potential pitfall as the CALR variant detected was novel with no known clinical significance, but is predicted to be a benign variant due to preservation of the reading frame and C-terminal KDEL signal sequence. Clearly communicating the type of CALR variant, or in our case a likely benign variant, will minimize the possibility of misdiagnosis and overtreatment. If a patient is suspected of harboring a germline CALR variant, it is recommended that a non-hematopoietic source (i.e. buccal swab) be obtained for result confirmation. Unfortunately, our patient has been lost to follow-up, precluding our ability to obtain a buccal swab for confirmation testing and obtain follow-up clinical outcomes data. However, assessment of the allele fraction (Fig. 2) is supportive of a germline heterozygous variant since the allele fraction is approximately 50 percent. We speculate that in the coming years, additional patients with coexisting JAK2 V617 and CALR variants will be discovered and the clinical significance, prognosis, and outcomes of these patients will be further defined.
- Swerdlow SH, Campo E, Harris NL, et al., eds. WHO Classification of Tumours of Haematopoietic and Lymphoid Tissues. Rev. 4th ed.; vol. 2. Lyon, France: IARC; 2017.
- Barbui T, Thiele J, Vannucchi AM, Tefferi A. Rationale for revision and proposed changes of the WHO diagnostic criteria for polycythemia vera, essential thrombocythemia and primary myelofibrosis. Blood Cancer J. 2015;5:e337.
- Rashid M, Ahmed RZ, Ahmed S, Nadeem M, Ahmed N, Shamsi TS. Coexisting JAK2V617F and CALR exon 9 mutation in essential thrombocythemia. Indian J Hematol Blood Transfus. 2016;32(suppl 1):112–116.
- Lim KH, Chang YC, Gon-Shen Chen C, et al. Frequent CALR exon 9 alterations in JAK2 V617F-mutated essential thrombocythemia detected by high-resolution melting analysis. Blood Cancer J. 2015;5:e295.
- Klampfl T, Gisslinger H, Harutyunyan AS, et al. Somatic mutations of calreticulin in myeloproliferative neoplasms. N Engl J Med. 2013;369(25):2379–2390.
- Nangalia J, Massie CE, Baxter EJ, et al. Somatic CALR mutations in myeloproliferative neoplasms with nonmutated JAK2. N Engl J Med. 2013;369(25):2391–2405.
- Chachoua I, Pecquet C, El-Khoury M, et al. Thrombopoietin receptor activation by myeloproliferative neoplasm associated calreticulin mutants. Blood. 2016;127(10):1325–1335.
- Szuber N, Lamontagne B, Busque L. Novel germline mutations in the calreticulin gene: implications for the diagnosis of myeloproliferative neoplasms. J Clin Pathol. 2016;69(11):1033–1036.
- McGaffin G, Harper K, Stirling D, McLintock L. JAK2 V617F and CALR mutations are not mutually exclusive; findings from retrospective analysis of a small patient cohort. Br J Haematol. 2014;167(2):276–278.
- Xu N, Ding L, Yin C, et al. A report on the co-occurrence of JAK2V617F and CALR mutations in myeloproliferative neoplasm patients. Ann Hematol. 2015;94(5):865–867.
- Murugesan G, Guenther-Johnson J, Mularo F, Cook JR, Daly TM. Validation of a molecular diagnostic assay for CALR exon 9 indels in myeloproliferative neoplasms: identification of coexisting JAK2 and CALR mutations and a novel 9 bp deletion in CALR. Int J Lab Hematol. 2016;38(3):284–297.
- Al Assaf C, Van Obbergh F, Billiet J, et al. Analysis of phenotype and outcome in essential thrombocythemia with CALR or JAK2 mutations. Haematologica. 2015;100(7):893–897.
Dr. Haberichter was (at the time of writing) a hematopathology fellow, Dr. Blenc is medical director of the hematology laboratory and director of the hematopathology fellowship program, and Anne Prada is a medical technologist II in the molecular pathology laboratory—all in the Department of Pathology and Laboratory Medicine, Beaumont Health, Royal Oak, Mich. Dr. Haberichter is now a staff hematopathologist, Grand Traverse Pathology, Traverse City, Mich. All content and writing of the manuscript were completed while Dr. Boyanton was medical director of the microbiology and molecular pathology laboratories in the Department of Pathology and Laboratory Medicine, Beaumont Health. He is now professor of pathology, University of Arkansas for Medical Sciences, and pathologist-in-chief, Arkansas Children’s Hospital, Little Rock.
Test yourself
[tabs type=”horizontal”][tabs_head][tab_title]Here are three questions taken from the case report.[/tab_title][tab_title]Answers[/tab_title][/tabs_head][tab]Answers are online at www.amp.org/casereports and will be published next month in CAP TODAY.
1. Which of the following statements is true regarding somatic CALR variants?
a. Variants are found in less than one percent of patients with essential thrombocythemia.
b. JAK2 V617F and CALR variants are mutually exclusive.
c. These are frameshift variants that interrupt the reading frame.
d. Patients have a more aggressive clinical course and increased thrombotic risk.
2. Which of the following would be considered a germline CALR variant?
a. 52-basepair deletion
b. 5-basepair insertion
c. 9-basepair deletion
d. 1-basepair deletion
3. Which of the following is true regarding germline CALR variants?
a. As the sole detected variant, they are not associated with myeloproliferative neoplasms.
b. They may be seen in conjunction with somatic JAK2 V617F or somatic CALR variants.
c. These are in-frame variants that preserve the reading frame.
d. All of the above.[/tab]
[tab]
1. Which of the following statements is true regarding somatic CALR variants?
a. Variants are found in less than one percent of patients with essential thrombocythemia.
b. JAK2 V617F and CALR variants are mutually exclusive.
c. These are frameshift variants that interrupt the reading frame.
d. Patients have a more aggressive clinical course and increased thrombotic risk.
2. Which of the following would be considered a germline CALR variant?
a. 52-basepair deletion
b. 5-basepair insertion
c. 9-basepair deletion
d. 1-basepair deletion
3. Which of the following is true regarding germline CALR variants?
a. As the sole detected variant, they are not associated with myeloproliferative neoplasms.
b. They may be seen in conjunction with somatic JAK2 V617F or somatic CALR variants.
c. These are in-frame variants that preserve the reading frame.
d. All of the above.
[/tab][/tabs]