
![]() CAP TODAY and the Association for Molecular Pathology have teamed up to bring molecular case reports to CAP TODAY readers. AMP members write the reports using clinical cases from their own practices that show molecular testing’s important role in diagnosis, prognosis, and treatment. The following report comes from Children’s Hospital of Philadelphia and Driscoll Children’s Hospital, Corpus Christi, Tex. If you would like to submit a case report, please send an email to the AMP at amp@amp.org. For more information about the AMP and all previously published case reports, visit www.amp.org.
CAP TODAY and the Association for Molecular Pathology have teamed up to bring molecular case reports to CAP TODAY readers. AMP members write the reports using clinical cases from their own practices that show molecular testing’s important role in diagnosis, prognosis, and treatment. The following report comes from Children’s Hospital of Philadelphia and Driscoll Children’s Hospital, Corpus Christi, Tex. If you would like to submit a case report, please send an email to the AMP at amp@amp.org. For more information about the AMP and all previously published case reports, visit www.amp.org.
Jinhua Wu, PhD; Jeffrey Schubert, PhD
Xiaonan Zhao, PhD; Elizabeth Fanning, MS
Zhiqian Fan, MS; Lisa Sutton, MD; Marilyn M. Li, MD
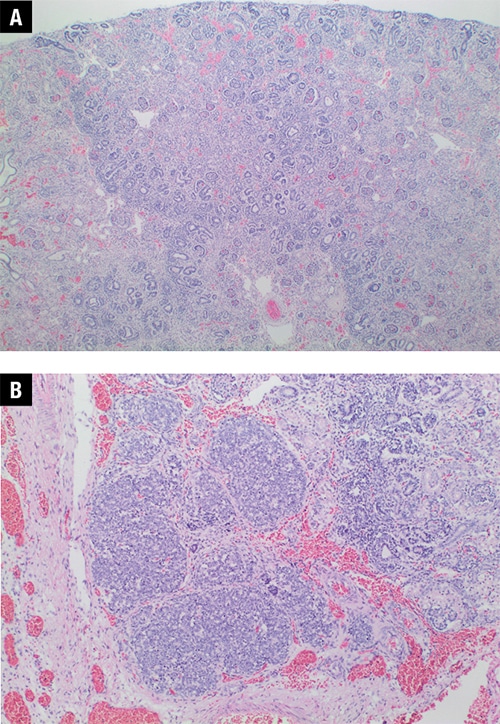
December 2019—We report a case of a 34-week-gestation male infant with prenatal overgrowth born to a 19-year-old G1P0 mother. No significant family history or consanguinity were reported. The infant was delivered emergently by cesarean section due to pregnancy-induced hypertension and polyhydramnios. At birth, the infant was noted to have anasarca and ascites with a massively distended abdomen and enlarged kidneys. Despite resuscitative efforts, the infant expired at one day of life. A complete autopsy demonstrated clinical and histologic features suggestive of Perlman syndrome. There were significant urinary tract abnormalities, including massive nephromegaly, hydronephrosis with megaureters, bilateral diffuse nephroblastomatosis, and renal dysplastic changes (Fig. 1). Also noted was organomegaly of the liver, spleen, and thymus. The lungs showed mild pulmonary hypoplasia with diffuse hyaline membrane disease. A skin biopsy was sent to our laboratory for genetic testing with suspicion of Perlman syndrome (mutation in DIS3L2 gene) or other potential causes for diseases with elevated cancer risk in an overgrown fetus, such as Simpson-Golabi-Behmel syndrome (GPC3). For these reasons, a comprehensive hereditary cancer panel of 130 genes (including DIS3L2) was performed.
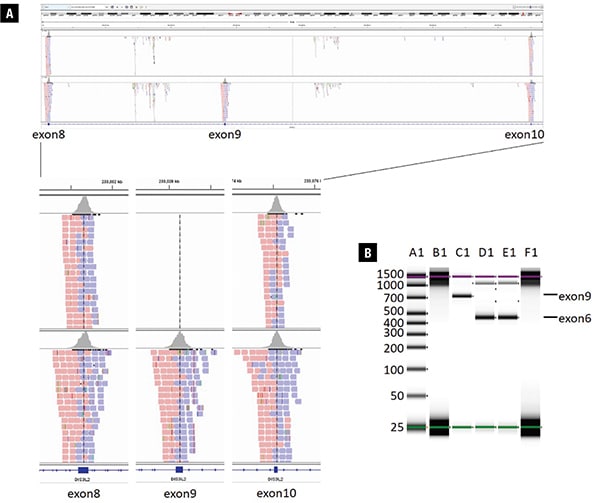
The comprehensive hereditary cancer panel did not identify a pathogenic or likely pathogenic variant according to the American College of Medical Genetics and Genomics/Association for Molecular Pathology germline variant classification guidelines,1 and no variants were observed in the DIS3L2 gene. However, analysis of regions with low sequence coverage on the panel discovered no sequence reads aligned to the genomic region of chr2:233028163-233028348, corresponding to exon 9 of DIS3L2 (NM_152383.4), indicating a homozygous deletion of DIS3L2 exon 9 as visualized using Integrative Genomics Viewer (IGV) (Fig. 2A). The deletion was confirmed by PCR specifically targeting DIS3L2 exon 9 using exon 6 as a positive PCR control (Fig. 2B). Precise breakpoints could not be determined due to the nature of the targeted assay used. Homozygous deletions of exon 9 in DIS3L2 have been reported in multiple individuals from at least four families with Perlman syndrome2-4 and are listed in DECIPHER (#318416, https://decipher.sanger.ac.uk/). The deletion of exon 9 is predicted to result in an in-frame mRNA product, and it may be mediated by nonallelic homologous recombination between LINE-1 repeats located in the introns.3 The identification of the DIS3L2 exon 9 homozygous deletion is diagnostic of Perlman syndrome for the patient.
Next-generation sequencing technology allows interrogation of multiple genes in parallel to evaluate multiple variant types, including SNVs, small indels, exonic deletions/duplications, and gross copy number changes, as well as fusions and alternative splicing, through a single test, using DNA and RNA. PCR was used to confirm the exon 9 homozygous deletion in this case since we knew what we were looking for; however, this method would have had limited utility as a first-line test because it cannot detect the same spectrum of variants in as many genes as can NGS-based tests.
Perlman syndrome, first described in the 1970s,5-7 is a rare, autosomal recessive overgrowth syndrome that can present prenatally with fetal macrosomia and polyhydramnios. Patients also show characteristic dysmorphic facial features, including deep-set eyes, low-set ears, inverted V-shaped upper lip, prominent forehead, and a broad, flat nasal bridge. There is a high rate of neonatal mortality in patients with Perlman syndrome. Those who do survive commonly exhibit developmental delay and renal abnormalities and are at a high risk for developing nephroblastomatosis and Wilms tumor at an early age.8,9
Astuti, et al., established a molecular basis for Perlman syndrome in 20122 by identifying regions of homozygosity in consanguineous Perlman syndrome families. With further genetic mapping, germline homozygous deletions of DIS3L2 exons were found in multiple consanguineous families, and additional biallelic DIS3L2 single nucleotide variants, including splice-site and missense mutations, were found to segregate with disease in non-consanguineous patients. The authors further demonstrated through functional in vitro studies that recombinant DIS3L2 protein with exon 9 homozygous deletion displayed substantially reduced ribonuclease activity compared with wild-type DIS3L2. Additionally, loss of this gene in cell lines led to increased rates of aneuploidy, errors in mitosis, and abnormal expression of mitotic proteins.
In this case, a confirmed molecular diagnosis of Perlman syndrome has two primary impacts on patient care. First, in a situation of a neonatal death, a definitive diagnosis can help bring closure to the pregnancy for this family by providing a direct answer to what caused their loss. Second, and most significantly, this diagnosis has important clinical implications for this couple’s family planning and future pregnancies. As part of the report issued in this case, we recommended genetic testing for this family to aid in the assessment of recurrence risk. If both parents are found to be asymptomatic carriers for heterozygous pathogenic variants in DIS3L2, there is a 25 percent chance of their having another child with Perlman syndrome and a 50 percent chance of their having a child who is a carrier.
In sum, we identified a homozygous deletion of a single exon of DIS3L2 gene using an NGS-based hereditary cancer panel in a patient who died soon after birth. This result not only provides a molecular diagnosis of Perlman syndrome but also carries important implications for the family in its decision-making for future pregnancies. Our experience demonstrates that an NGS-based assay can detect copy number variants at the single exon level through use of a carefully designed bioinformatics pipeline and thorough sequence data investigation.
- Richards S, Aziz N, Bale S, et al. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med. 2015;17(5):405–424.
- Astuti D, Morris MR, Cooper WN, et al. Germline mutations in DIS3L2 cause the Perlman syndrome of overgrowth and Wilms tumor susceptibility. Nat Genet. 2012;44(3):277–284.
- Higashimoto K, Maeda T, Okada J, et al. Homozygous deletion of DIS3L2 exon 9 due to non-allelic homologous recombination between LINE-1s in a Japanese patient with Perlman syndrome. Eur J Hum Genet. 2013;21(11):1316–1319.
- Henneveld HT, van Lingen RA, Hamel BC, Stolte-Dijkstra I, van Essen AJ. Perlman syndrome: four additional cases and review. Am J Med Genet. 1999;86(5):439–446.
- Liban E, Kozenitzky IL. Metanephric hamartomas and nephroblastomatosis in siblings. Cancer. 1970;25(4):885–888.
- Perlman M, Goldberg GM, Bar-Ziv J, Danovitch G. Renal hamartomas and nephroblastomatosis with fetal gigantism: a familial syndrome. J Pediatr. 1973;83(3):414–418.
- Perlman M, Levin M, Wittels B. Syndrome of fetal gigantism, renal hamartomas, and nephroblastomatosis with Wilms’ tumor. Cancer. 1975;35(4):1212–1217.
- Alessandri JL, Cuillier F, Ramful D, et al. Perlman syndrome: report, prenatal findings and review. Am J Med Genet A. 2008;146A(19):2532–2537.
- Perlman EJ. Pediatric renal tumors: practical updates for the pathologist. Pediatr Dev Pathol. 2005;8(3):320–338.
Dr. Wu and Dr. Schubert are clinical genomic scientists, Dr. Zhao is genomic section lead scientist, Fanning is NGS analyst, Fan is clinical laboratory section supervisor, and Dr. Li is vice chief—all in the Division of Genomic Diagnostics, Department of Pathology and Laboratory Medicine, Children’s Hospital of Philadelphia. Dr. Sutton is a pediatric pathologist, Department of Pathology and Laboratory Medicine, Driscoll Children’s Hospital, Corpus Christi, Tex.
Test yourself
[tabs type=”horizontal”][tabs_head][tab_title]Here are three questions taken from the case report.[/tab_title][tab_title]Answers[/tab_title][/tabs_head][tab]Answers are online at www.amp.org/casereports and will be published next month in CAP TODAY.
1. What sequencing technology was used in this case report? What types of variants can be detected by this method?
a. Next-generation sequencing. It detects single nucleotide variants, small deletions and insertions, and copy number variation (DNA), and it can also detect alternative splicing and gene fusions (RNA).
b. Sanger sequencing. It detects single nucleotide variants, small deletions and insertions, and copy number variations.
c. Third-generation sequencing using Nanopore technology. It detects single nucleotide variants and small deletions and insertions. It does not detect copy number variations.
d. Sanger sequencing. It detects single nucleotide variants only.
2. What is Perlman syndrome? A mutation in what gene causes this disease?
a. Perlman syndrome is a congenital overgrowth syndrome inherited in an autosomal recessive manner that is associated with Wilms tumor susceptibility. DIS3L1 and DIS3L2.
b. Perlman syndrome is a genetic disorder due to loss of function of specific genes. In newborns, symptoms include weak muscles, poor feeding, and slow development. DIS3L2.
c. Perlman syndrome is a congenital overgrowth syndrome inherited in an autosomal recessive manner that is associated with Wilms tumor susceptibility. DIS3L2.
d. Perlman syndrome is a genetic disorder due to loss of function of specific genes. In newborns, symptoms include weak muscles, poor feeding, and slow development. RET.
3. What is the inheritance pattern of Perlman syndrome? What is the recurrence risk of having another child with Perlman syndrome in this family?
a. It follows an autosomal recessive pattern of inheritance. Therefore, there is a 25 percent chance of the couple having another child with Perlman syndrome and a 50 percent chance of having a child who is a carrier.
b. It follows an autosomal dominant pattern of inheritance. Since only the proband was tested in this family, parents should be tested for this variant as well to determine whether this variant is inherited or de novo.
c. It follows an autosomal recessive or autosomal dominant pattern of inheritance. The risk for another child with Perlman syndrome is 100 percent for this family.
d. It follows an autosomal recessive pattern of inheritance. Since only the proband was tested in this family, parents should be tested for this variant as well to determine if both parents are a carrier of this deletion.
[/tab]
[tab]
1. What sequencing technology was used in this case report? What types of variants can be detected by this method?
a. Next-generation sequencing. It detects single nucleotide variants, small deletions and insertions, and copy number variation (DNA), and it can also detect alternative splicing and gene fusions (RNA).
b. Sanger sequencing. It detects single nucleotide variants, small deletions and insertions, and copy number variations.
c. Third-generation sequencing using Nanopore technology. It detects single nucleotide variants and small deletions and insertions. It does not detect copy number variations.
d. Sanger sequencing. It detects single nucleotide variants only.
2. What is Perlman syndrome? A mutation in what gene causes this disease?
a. Perlman syndrome is a congenital overgrowth syndrome inherited in an autosomal recessive manner that is associated with Wilms tumor susceptibility. DIS3L1 and DIS3L2.
b. Perlman syndrome is a genetic disorder due to loss of function of specific genes. In newborns, symptoms include weak muscles, poor feeding, and slow development. DIS3L2.
c. Perlman syndrome is a congenital overgrowth syndrome inherited in an autosomal recessive manner that is associated with Wilms tumor susceptibility. DIS3L2.
d. Perlman syndrome is a genetic disorder due to loss of function of specific genes. In newborns, symptoms include weak muscles, poor feeding, and slow development. RET.
3. What is the inheritance pattern of Perlman syndrome? What is the recurrence risk of having another child with Perlman syndrome in this family?
a. It follows an autosomal recessive pattern of inheritance. Therefore, there is a 25 percent chance of the couple having another child with Perlman syndrome and a 50 percent chance of having a child who is a carrier.
b. It follows an autosomal dominant pattern of inheritance. Since only the proband was tested in this family, parents should be tested for this variant as well to determine whether this variant is inherited or de novo.
c. It follows an autosomal recessive or autosomal dominant pattern of inheritance. The risk for another child with Perlman syndrome is 100 percent for this family.
d. It follows an autosomal recessive pattern of inheritance. Since only the proband was tested in this family, parents should be tested for this variant as well to determine if both parents are a carrier of this deletion.
[/tab]
[/tabs]