
CAP TODAY and the Association for Molecular Pathology have teamed up to bring molecular case reports to CAP TODAY readers. AMP members write the reports using clinical cases from their own practices that show molecular testing’s important role in diagnosis, prognosis, and treatment. The following report comes from Hospital of the University of Pennsylvania. If you would like to submit a case report, please send an email to the AMP at amp@amp.org. For more information about the AMP and all previously published case reports, visit www.amp.org.
Narek Israelyan, MD, MS
Dylane Wineland, MS, LCGC
Salvatore F. Priore, MD, PhD
Jacquelyn J. Roth, PhD
October 2023—Next-generation sequencing of tumor tissue has important implications in solid and hematologic malignancies because it can identify genomic variants that provide diagnostic, prognostic, and predictive information to guide clinical management. Variants identified on tumor sequencing can be classified as somatic (acquired after conception) or inherited through germline. Identification of germline variants can alter the treatment of current tumors, as in the case of BRCA-associated breast and ovarian cancers1; provide prognostic information about the risk of developing future cancer; and have preventive value by suggesting screening and follow-up for relatives. Guidelines from the American Society of Clinical Oncology recommend communicating to patients medically relevant incidental germline findings from somatic mutation profiling conducted in the clinical setting,2 and the American College of Medical Genetics and Genomics has established a minimum list for reporting secondary findings from clinical exome and genome sequencing.3 Here we present a case with unexpected findings of pathogenic variants in genes not related to the indication for sequencing, highlighting the importance of a workflow for specialized follow-up of clinically relevant potential germline variants.
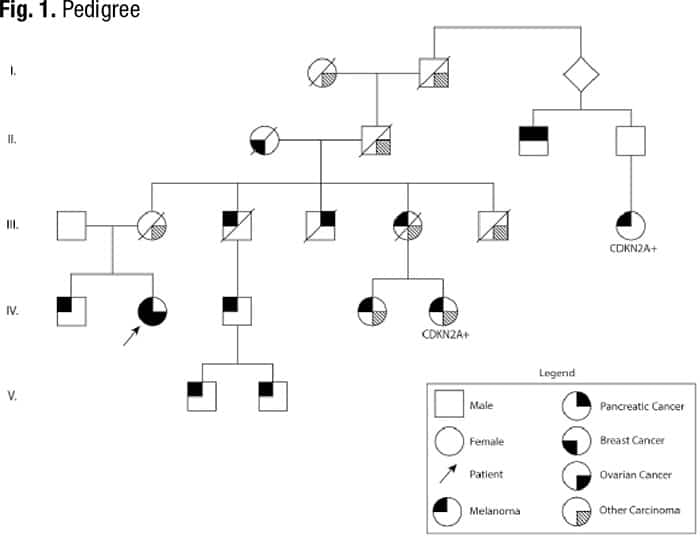
Case. A 71-year-old female patient with a history of breast cancer and melanoma presented to her hematologist for follow-up of ongoing thrombocytopenia and new onset postmenopausal bleeding. She was diagnosed at ages 49, 51, and 68 with malignant melanoma on the right breast and back, which were managed with surgical resection. The patient was diagnosed at age 60 with grade three ductal carcinoma in situ (ER+, PR+, HER2+) and underwent lumpectomy with adjunctive radiation and five years of tamoxifen. She reports a strong family history of cancer, including melanoma in both first- and second-degree relatives, and history of a pathogenic germline CDKN2A variant in a third-degree relative, but she has not completed genetic counseling or germline testing (Fig. 1).
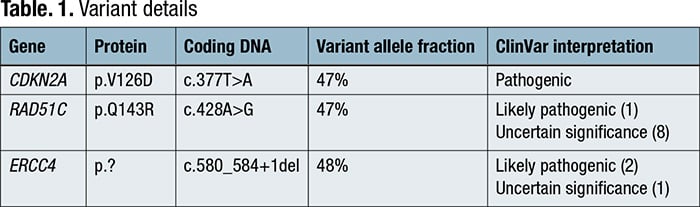
Complete blood count revealed a low platelet count of 59,000/μL, consistent over six months, and a mild macrocytosis in an otherwise normal CBC. Coagulation testing was normal. Bone marrow aspiration and biopsy were performed, which showed a mildly hypercellular marrow (40 percent) with trilineage hematopoiesis and no overt evidence of a hematolymphoid neoplasm. Cancer cytogenetic evaluation revealed a normal karyotype (46,XX[20]). Flow cytometry showed no immunophenotypic evidence of a hematolymphoid neoplasm. Next-generation sequencing was performed on the bone marrow aspirate, which identified pathogenic variants in CDKN2A and ERCC4 and a probable pathogenic variant in RAD51C, all potentially germline based on variant allele frequencies (VAF) (Table 1). Following our institution’s algorithm for incidental germline findings, variants identified in CDKN2A and RAD51C triggered a message to the ordering clinical team recommending genetic counseling and follow-up germline genetic testing if clinically warranted.
For further evaluation of her postmenopausal bleeding, the patient underwent a transvaginal ultrasound that demonstrated an abnormal right ovary with a complex cystic and solid mass, measuring 9.7 × 8.5 × 11.9 cm on subsequent CT examination. Exploratory laparotomy and total abdominal hysterectomy were performed, with microscopic examination revealing stage IIIA2 high-grade serous carcinoma of bilateral ovaries, including involvement of peritoneum. The sample was sent for homologous recombination deficiency (HRD) testing, which resulted with a positive genomic instability score, indicating improved prognosis and potential responsiveness of the tumor to PARP inhibitors.4 BRCA1 and BRCA2 variants were not identified in this specimen.
The patient was ultimately referred to genetic counseling and proceeded with comprehensive germline testing, which included evaluation of genes related to hereditary breast and ovarian cancer syndromes. Germline testing confirmed the presence of CDKN2A and RAD51C variants that were found on somatic testing. ERCC4 was not included in the germline panel because her personal and family history was not suggestive of ERCC4-related hereditary syndromes.
Discussion. A minority of patients referred for molecular tumor tissue analysis may harbor germline variants associated with hereditary cancer syndromes. While some labs perform tumor-normal paired testing to separate germline from somatic variants, most perform tumor-only testing, which requires a thoughtful plan for identifying and referring patients with suspected pathogenic germline variants. These referral pipelines are currently institution-specific5; however, the recently published ACMG SF v3.1 guidelines list germline variants that, when found incidentally, warrant informing patients and referring them for genetic counseling.3 The patient described, with a strong family history of cancer and personal history of breast cancer and melanoma, had somatic testing done on a bone marrow sample that demonstrated no overt evidence of a hematolymphoid neoplasm. Sequencing performed on this sample incidentally uncovered variants in CDKN2A, RAD51C, and ERCC4 that were likely germline due to a near 50 percent variant allele fraction, some of which are associated with hereditary cancer syndromes.
Cyclin-dependent kinase inhibitor 2A (CDKN2A) is a tumor suppressor gene located on chromosome 9p21.3 and encodes two proteins: p16-INK4A and p14-ARF. These proteins promote cell cycle arrest or apoptosis.6,7 CDKN2A is the most commonly mutated gene in hereditary melanoma, found in 20 to 40 percent of high-risk families, and is associated with a worse overall survival from both melanoma and nonmelanoma cancers.8,9 Familial melanomas with CDKN2A mutations have a unique histologic phenotype of dense pigmentation, high pagetoid scatter, and a non-spindle cell morphology in the vertical growth phase.10 The CDKN2A p.V126D mutation identified in our patient has been previously observed in the germline of patients with familial melanoma and pancreatic cancer.11 Germline CDKN2A mutations have also been strongly associated with development of familial pancreatic cancer and less commonly with head and neck carcinomas, breast carcinomas, nervous system tumors, sarcomas, and other carcinomas.12,13
The RAD51C gene located on chromosome 17q22 is a protein involved in double-strand break repair and interstrand crosslink repair by homologous recombination.14 Germline mutations in RAD51C are associated with Fanconi anemia-like disorders in addition to hereditary breast and ovarian cancers.15-17 RAD51C was not included in the ACMG SF v3.1 list for reporting of secondary findings in clinical sequencing on the basis of penetrance considerations and absence of effective ovarian cancer screening.3 Despite this, recent large population-based studies have reported that pathogenic germline variants in RAD51C are associated with an increased risk of ER-negative breast cancer in both female and male patients, with a risk of up to 30 percent, particularly with truncating variants.18,19
The excision repair 4, endonuclease catalytic subunit (ERCC4) gene, also known as FANCQ and XPF, is located on chromosome 16p13.12. It encodes a subunit for the ERCC1-XPF enzyme complex that is a core component of nucleotide excision repair and participates in double-stand break repair and interstrand crosslink repair by homologous recombination.20 ERCC4 germline mutations have been identified in Fanconi anemia, xeroderma pigmentosa, XFE progeroid syndrome, and Cockayne syndrome, and in a small subset of hereditary breast cancers.21,22
With patients who harbor potential germline variants, we follow institution-specific guidelines. These guidelines include VAF-independent referrals, given that genetic changes like loss of heterozygosity in tumors can alter VAF, and referrals for genes that may or may not be involved in the somatic indication for sequencing. Some referrals may be age based, including those for APC, RB1, NF1, and TP53 in individuals with cancer diagnosed under the age of 30. Known germline founder variants are automatically referred. In addition, referrals may be based on phasing of mutations, for example, only referring biallelic MUTYH. Lastly, referrals often depend on prior treatments, including only referring EGFR T790M in the setting of EGFR-treatment naïve patients.
After identifying these potentially actionable germline variants and obtaining the results of homologous recombination deficiency testing, the patient was deemed a candidate for PARP inhibitors as adjunctive therapy for ovarian cancer. Due to the strong relationship of CDKN2A variants with familial melanoma and pancreatic cancer syndromes, the benefit of predictive genetic testing in relatives was discussed with the patient, as the patient has two nephews with melanoma but without known variants. While the patient has no offspring, she has one full sibling for whom predictive genetic testing was recommended. She was referred for consideration of pancreatic cancer screening studies through contrast-enhanced MRI/magnetic resonance cholangiopancreatography and/or endoscopic ultrasound and will continue with semiannual skin exams with dermatology. She will also continue with annual mammograms for breast screening. Multiple commercial genetic testing labs currently classify the detected RAD51C variant as a variant of uncertain significance, although there is conflicting data for pathogenicity. In addition, the germline status and clinical implications of the ERCC4 variant is still unknown. Providers who are familiar with hereditary cancer syndromes will provide the follow-up care because current knowledge surrounding these germline variants continues to evolve and may have an impact on future management.
- Lee J-M, Ledermann JA, Kohn EC. PARP inhibitors for BRCA1/2 mutation-associated and BRCA-like malignancies. Ann Oncol. 2014;25(1):32–40.
- Robson ME, Bradbury AR, Arun B, et al. American Society of Clinical Oncology policy statement update: genetic and genomic testing for cancer susceptibility. J Clin Oncol. 2015;33(31):3660–3667.
- Miller DT, Lee K, Abul-Husn NS, et al; ACMG Secondary Findings Working Group. ACMG SF v3.1 list for reporting of secondary findings in clinical exome and genome sequencing: a policy statement of the American College of Medical Genetics and Genomics (ACMG). Genet Med. 2022;24(7):1407–1414.
- Ledermann JA. PARP inhibitors in ovarian cancer. Ann Oncol. 2016;27(suppl 1):i40–i44.
- Clark DF, Maxwell KN, Powers J, et al. Identification and confirmation of potentially actionable germline mutations in tumor-only genomic sequencing. JCO Precis Oncol. 2019;3. Published online Aug. 19, 2019. doi:10.1200/PO.19.00076
- Li J, Poi MJ, Tsai MD. Regulatory mechanisms of tumor suppressor P16INK4A and their relevance to cancer. Biochemistry. 2011;50(25):5566–5582.
- Weber HO, Samuel T, Rauch P, Funk JO. Human p14ARF-mediated cell cycle arrest strictly depends on intact p53 signaling pathways. Oncogene. 2002;21(20):3207–3212.
- Rossi M, Pellegrini C, Cardelli L, Ciciarelli V, Di Nardo L, Fargnoli MC. Familial melanoma: diagnostic and management implications. Dermatol Pract Concept. 2019;9(1):10–16.
- Helgadottir H, Höiom V, Tuominen R, et al. Germline CDKN2A mutation status and survival in familial melanoma cases. J Natl Cancer Inst. 2016;108(11):djw135.
- Sargen MR, Kanetksy PA, Newton-Bishop J, et al. Histologic features of melanoma associated with CDKN2A genotype. J Am Acad Dermatol. 2015;72(3):P496-507.E7.
- Hussussian CJ, Struewing JP, Goldstein AM, et al. Germline p16 mutations in familial melanoma. Nat Genet. 1994;8(1):15–21.
- Bartsch DK, Sina-Frey M, Lang S, et al. CDKN2A germline mutations in familial pancreatic cancer. Ann Surg. 2002;236(6):730–737.
- Chan SH, Chiang J, Ngeow J. CDKN2A germline alterations and the relevance of genotype-phenotype associations in cancer predisposition. Hered Cancer Clin Pract. 2021;19(1):21.
- Prakash R, Rawal Y, Sullivan MR, et al. Homologous recombination-deficient mutation cluster in tumor suppressor RAD51C identified by comprehensive analysis of cancer variants. Proc Natl Acad Sci U S A. 2022;119(38):e2202727119.
- Somyajit K, Subramanya S, Nagaraju G. RAD51C: a novel cancer susceptibility gene is linked to Fanconi anemia and breast cancer. Carcinogenesis. 2010;31(12):2031–2038.
- Clague J, Wilhoite G, Adamson A, Bialis A, Weitzel JN, Neuhausen SL. RAD51C germline mutations in breast and ovarian cancer cases from high-risk families. PLoS One. 2011;6(9):e25632.
- Rizzolo P, Zelli V, Silvestri V, et al. Insight into genetic susceptibility to male breast cancer by multigene panel testing: results from a multicenter study in Italy. Int J Cancer. 2019;145(2):390–400.
- Breast Cancer Association Consortium; Dorling L, Carvahlo S, Allen J, et al. Breast cancer risk genes—association analysis in more than 113,000 women. N Engl J Med. 2021;384(5):428–439.
- Hu C, Hart SN, Gnanaolivu R, et al. A population-based study of genes previously implicated in breast cancer. N Engl J Med. 2021;384(5):440–451.
- Manandhar M, Boulware KS, Wood RD. The ERCC1 and ERCC4 (XPF) genes and gene products. Gene. 2015;569(2):153–161.
- Kashiyama K, Nakazawa Y, Pilz DT, et al. Malfunction of nuclease ERCC1-XPF results in diverse clinical manifestations and causes Cockayne syndrome, xeroderma pigmentosum, and Fanconi anemia. Am J Hum Genet. 2013;92(5):807–819.
- Kohlhase S, Bogdanova NV, Schürmann P, et al. Mutation analysis of the ERCC4/FANCQ gene in hereditary breast cancer. PLoS One. 2014;9(1):e85334.
Dr. Israelyan is a third-year pathology resident in clinical pathology, Dylane Wineland is a genetic counselor, Dr. Priore is assistant professor of clinical pathology and laboratory medicine, and Dr. Roth is assistant professor of clinical pathology and laboratory medicine—all at the Hospital of the University of Pennsylvania and the Perelman School of Medicine, Philadelphia.
Test yourself
Here are three questions taken from the case report. Answers are online now at www.amp.org/casereports and will be published next month in CAP TODAY.
1. Which of the following biomarker descriptor terms best defines the role of homologous recombination deficiency testing in hereditary breast and ovarian cancer syndromes?
a. Diagnostic
b. Prognostic
c. Predictive
d. Diagnostic and prognostic
e. Prognostic and predictive
2. Germline alterations in CDKN2A are associated with predisposition to which of the following hereditary cancer syndromes?
a. Hereditary breast and ovarian cancer syndrome
b. Lynch syndrome (hereditary nonpolyposis colorectal cancer)
c. Melanoma-pancreatic cancer syndrome
d. Li-Fraumeni syndrome
e. Cowden syndrome
3. Which of the following suggestions are good practices with regard to genetic counseling referrals in patients with incidentally found germline variants on somatic-only testing?
a. Refer only if variant allele frequency is close to 50 percent
b. Refer regardless of whether germline mutation is related to somatic testing indication
c. Refer for APC, RB1, NF1, or TP53 variants regardless of patient age
d. Founder mutations do not need to be automatically referred
e. Refer MUTYH variants regardless of phasing
‘Test yourself’ answers
In the September 2023 issue was a case report, “Lung micropapillary adenocarcinomas revisited: A tale of antithesis with yearslong accumulative genetic alterations,” written by members of the Association for Molecular Pathology. Here are answers (in bold) to the three “test yourself ” questions that followed that case report.
1. Which of the following is the most common mutated gene in lung cancers?
a. KRAS
b. TP53
c. BRAF
d. EGFR
2. Which of the following statements about lung adenocarcinomas is false?
a. Micropapillary pattern of adenocarcinomas (MPC) are not considered aggressive variants of lung adenocarcinomas.
b. The presence of a micropapillary component of greater than five percent is an independent risk factor for recurrence.
c. The adverse prognosis of MPC is directly proportional to the percentage of micropapillary component.
d. Patients with MPC have lower five-year survival in stage-matched patients compared with other morphological variants.
3. Which of the following statements about lung carcinomas is false?
a. MPC is not the result of expanded growth of the papillary component.
b. TTF-1 immunohistochemistry positivity is seen in lung adenocarcinomas.
c. The most frequent EGFR gene mutations are targetable for therapy.
d. Newer mutations are acquired during tumor evolution.