
Neng Chen, PhD, DABMGG
Cathi Rubin Franklin, MS, LCGC, DABMGG
Franklin Quan, PhD, DABMGG
![]()
February 2018—CAP TODAY and the Association for Molecular Pathology have teamed up to bring molecular case reports to CAP TODAY readers. AMP members write the reports using clinical cases from their own practices that show molecular testing’s important role in diagnosis, prognosis, and treatment. The following report comes from Quest Diagnostics. If you would like to submit a case report, please send an email to the AMP at amp@amp.org. For more information about the AMP and all previously published case reports, visit www.amp.org.
The alpha-globin gene locus (Fig. 1A) contains two alpha-globin genes: HBA1 and HBA2. An upstream regulatory region, known as HS-40, is essential for alpha-globin gene expression.1,2 The loss or inactivation of one or two alpha-globin genes is not clinically significant while the loss or inactivation of all four alpha-globin genes results in a lethal condition known as hemoglobin (Hb) Barts hydrops fetalis syndrome, the most severe form of alpha-thalassemia. The loss or inactivation of three alpha-globin genes results in a form of alpha-thalassemia known as hemoglobin H (Hb H) disease.

Hb H disease has a variable phenotype. Hb H disease caused by deletions in the alpha-globin gene locus is associated with moderate anemia, microcytosis, and hypochromia. In contrast, nondeletional Hb H disease, caused by a point mutation or small insertion/deletion in one of the alpha-globin genes, is associated with a more severe anemia, and patients are likely to be transfusion dependent.
Patients with Hb H disease are often diagnosed after being seen for microcytic anemia or other indications, such as infection, hepatosplenomegaly, cholelithiasis, or severe anemia during pregnancy. For patients with low MCV, Heinz bodies after brilliant cresyl blue staining of blood smears, or Hb H and/or Hb Barts by high-performance liquid chromatography or gel electrophoresis, the differential diagnosis should include Hb H disease, hemolytic anemia, and alpha-thalassemia X-linked intellectual disability (ATRX) syndrome. Molecular genetic testing is used to confirm a diagnosis of Hb H disease and provide information for genetic counseling.
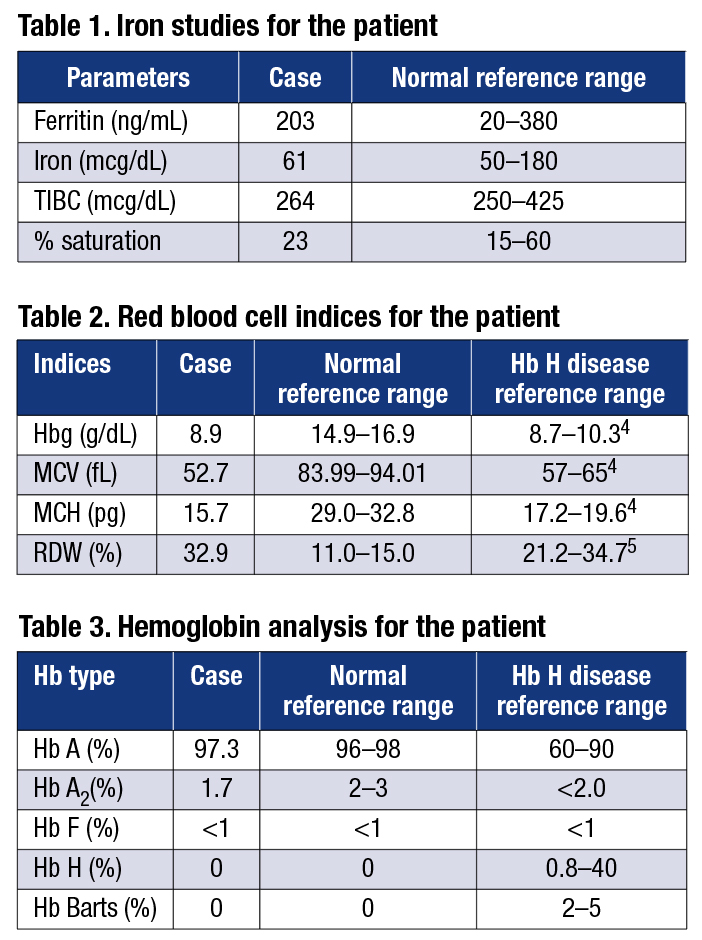
Patient case. A 50-year-old African-American male was evaluated by his primary care physician because of anemia. Several years prior, the patient had gastrointestinal bleeding and was diagnosed with colon cancer. He was anemic and transfused at that time as part of his treatment. His primary physician ordered blood smears and iron studies to investigate the cause of his anemia. The blood smears revealed microcytosis and the presence of target cells, indicating microcytic anemia. Iron studies indicated that all tested parameters were within the normal reference ranges (Table 1). Iron deficiency was ruled out as a cause of the patient’s anemia. The hemoccult test was positive at the time of his visit to the specialist.
The causes of anemia include severe iron deficiency, thalassemia, and chronic diseases (e.g. chronic infection or inflammatory processes).3 Anemia may also be idiopathic. The hematologist ordered a hemoglobinopathy evaluation (CBC and hemoglobin HPLC) to investigate the possibility of thalassemia as the cause of this patient’s anemia. The RBC indices (Table 2) clearly showed that this patient was severely anemic.
The hemoglobin HPLC (Table 3) was normal, with the exception of the Hb A2 level, which was slightly below the lower limit of the normal range. However, unstable Hb variants, such as Hb H, Hb Barts, or Hb Constant Spring (Hb CS), are not always detected by hemoglobin HPLC or gel electrophoresis. As a result, to rule out alpha-thalassemia as a cause of this patient’s anemia, molecular testing for mutations in the alpha-globin locus was performed.
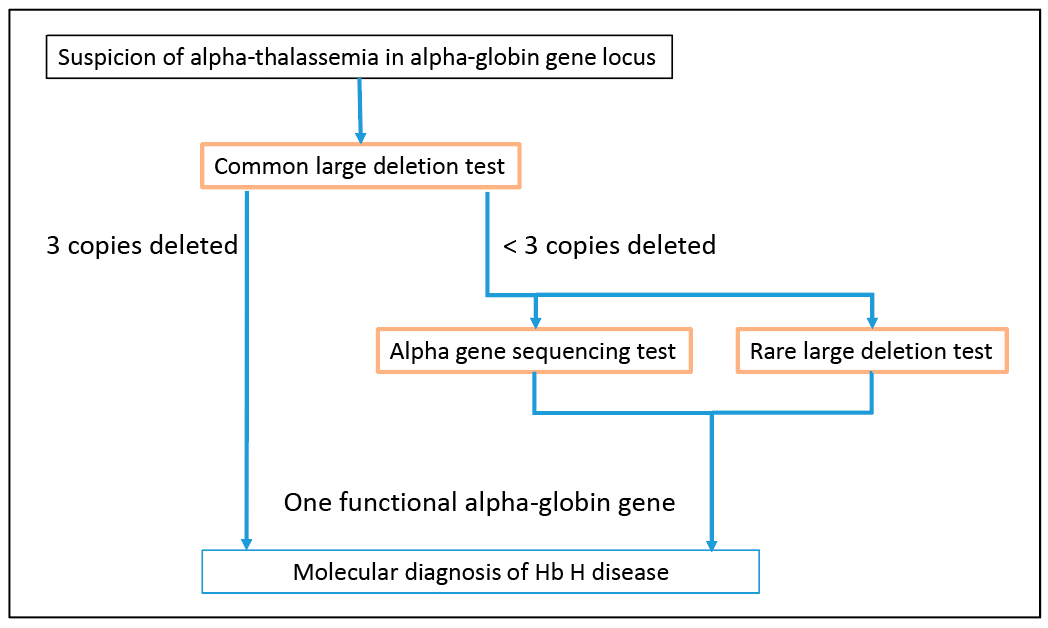
The most common mutations causing alpha-thalassemia are large deletions. Moreover, seven common deletions, -alpha3.7, -alpha4.2, –SEA, –MED, –FIL, –THAI, and –20.5,6 account for approximately 85 percent of all alpha-thalassemia mutations.7 Testing for these common deletions was performed using a multiplex gap-PCR assay.6 This testing indicated that the patient is homozygous for the -alpha3.7 deletion. The -alpha3.7 deletion generates a functional alpha-globin fusion gene that consists of exon 1 of the HBA2 gene and exons 2 and 3 of the HBA1 gene. Therefore, the test results indicated that the patient has two functional alpha-globin genes, a result that was inconsistent with his severe anemia.
To confirm that the patient is homozygous for the -alpha3.7 deletion (genotype -alpha3.7/-alpha3.7) and not heterozygous for the -alpha3.7 deletion and a rare, two alpha-globin gene deletion (genotype -alpha3.7/large deletion), alpha-globin gene dosage (i.e. copy number) was determined using a semiquantitative fluorescent PCR assay.8,9 This assay measures the relative dosage of amplification products derived from the HS-40 region, nonfunctional pseudogenes, and regions spanning the HBA2 and HBA1 genes (Fig. 1A). This assay confirmed that the patient is homozygous for the -alpha3.7 deletion. Unexpectedly, this assay also revealed the presence of a deletion that removes the HS-40 regulatory region on one chromosome. Due to the nature of the methodology used, the precise size and breakpoints of this deletion could not be determined. However, the data indicate that the deletion extends at least 11-kb upstream of the HS-40 region but not into the downstream pseudogene region (Fig. 1B).
The test results indicate that the patient has two alpha-globin fusion genes that should be functional. However, the HS-40 regulatory region is deleted on one chromosome. Since the HS-40 regulatory region is required for the expression of the alpha-globin genes on the same chromosome,1,2 the fusion gene on the chromosome with the HS-40 deletion is expected to be nonfunctional. Consequently, this patient has only one functional alpha-globin gene, which is consistent with a diagnosis of Hb H disease. Since deletions in the alpha-globin gene locus are the cause of this patient’s Hb H disease, he is expected to have the milder form of Hb H disease.4 As a result, it would not be surprising that his anemia was undiagnosed until he presented with GI bleeding and colon cancer. Unfortunately, we do not have information on the time course of his anemia.
Nonetheless, the RBC indices indicated this patient has anemia that is more severe than expected for an individual with the deletional form of Hb H disease (Table 2). The GI bleeding this patient experienced could cause iron deficiency, which would increase the severity of his anemia. However, adult patients with Hb H disease can have iron overload.4 Because this patient’s iron indices were in the normal range (Table 1), iron overload from Hb H disease could be compensating for the iron deficiency caused by GI bleeding. As a result, it is likely that both GI bleeding and Hb H disease have contributed to the severity of this patient’s microcytic anemia. Continued monitoring would be necessary to determine whether this individual’s anemia is less severe if the GI bleeding resolves. Genetic counseling and testing of at-risk family members are recommended.
 Discussion. It is straightforward to diagnose patients who have microcytic anemia that is caused by a single etiology. However, when there are multiple contributing factors, such as the GI bleeding and Hb H disease in our patient, interpreting the test results is more difficult. An iron study in isolation (Table 1) may be misleading because adult patients with Hb H disease can have iron overload. A comprehensive evaluation of iron studies, RBC indices, hemoglobin HPLC or gel electrophoresis, molecular testing, and clinical information is often necessary to arrive at the correct diagnosis and enable the selection of the appropriate management/treatment plan for the patient.
Discussion. It is straightforward to diagnose patients who have microcytic anemia that is caused by a single etiology. However, when there are multiple contributing factors, such as the GI bleeding and Hb H disease in our patient, interpreting the test results is more difficult. An iron study in isolation (Table 1) may be misleading because adult patients with Hb H disease can have iron overload. A comprehensive evaluation of iron studies, RBC indices, hemoglobin HPLC or gel electrophoresis, molecular testing, and clinical information is often necessary to arrive at the correct diagnosis and enable the selection of the appropriate management/treatment plan for the patient.
Large deletions are the most common molecular basis for alpha-thalassemia. The -alpha3.7, -alpha4.2, –SEA, –MED, –FIL, –THAI, and –20.5 deletions are the most common deletions causing alpha-thalassemia.6,7 As a result, testing for these deletions is usually the first-tier molecular test when alpha-thalassemia is suspected (Fig. 2). In our case, the first-tier testing indicated the patient is homozygous for the -alpha3.7 deletion. Additional testing for rare deletions resulted in the detection of a deletion involving the HS-40 regulatory region, which enabled the diagnosis of Hb H disease for this patient. Thus, it is necessary to perform additional testing for rare deletions and other types of mutations before excluding a diagnosis of Hb H disease.
In addition, when investigating the possibility that thalassemia is the cause of microcytic anemia, hemoglobin HPLC or gel electrophoresis provides clues to the etiology of the anemia by detecting abnormal hemoglobins. However, some unstable hemoglobin variants, such as Hb H, Hb Barts, and Hb CS, may be difficult to detect or not be detected by HPLC or gel electrophoresis. Molecular genetic analysis of the alpha- and/or beta-globin genes can be used to confirm the hemoglobin analysis findings. Therefore, molecular analysis is crucial to confirm or rule out thalassemia as a cause of anemia. Without molecular analysis, the diagnosis of Hb H disease would not have been made for this patient.
In summary, molecular analysis is critical in confirming the diagnosis of Hb H disease, although the diagnosis must be supported by the clinical presentation and other laboratory tests. Determining the genotype of an individual affected by Hb H disease using molecular methods provides information for genetic counseling and reproductive planning and assists in predicting the severity of the disease in affected individuals.
- Coelho A, Picanço I, Seuanes F, Seixas MT, Faustino P. Novel large deletions in the human alpha-globin gene cluster: clarifying the HS-40 long-range regulatory role in the native chromosome environment. Blood Cells Mol Dis. 2010;45(2):147–153.
- Sollaino MC, Paglietti ME, Loi D, Congiu R, Podda R, Galanello R. Homozygous deletion of the major alpha-globin regulatory element (MCS-R2) responsible for a severe case of hemoglobin H disease. Blood. 2010;
116(12):2193–2194. - Van Vranken M. Evaluation of microcytosis. Am Fam Physician. 2010;82(9):1117–1122.
- Chui DH, Fucharoen S, Chan V. Hemoglobin H disease: not necessarily a benign disorder. Blood. 2003;
101(3):791–800. - Venugopal S, Dhuri S, Al Jabal KB, Shaju A. Hemoglobin H disease in Muscat, Oman—a 5 year study. Oman Med J. 2008;23(2):82–85.
- Chong SS, Boehm CD, Higgs DR, Cutting GR. Single-tube multiplex-PCR screen for common deletional determinants of alpha-thalassemia. Blood. 2000;95(1):360–362.
- Origa R, Moi P. Alpha-Thalassemia. Nov. 1, 2005. Updated Dec. 29, 2016. In: Adam MP, Ardinger HH, Pagon RA, et al., eds. GeneReviews [database online]. Seattle: University of Washington; 1993–2017. www.ncbi.nlm.nih.gov/books/NBK1435/.
- Liu ET, He M, Rajgopal U. Differential polymerase chain reaction in the analysis of gene dosage. Semin Cancer Biol. 1993;4(1):47–58.
- Hantash FM, Redman JB, Starn K, et al. Novel and recurrent rearrangements in the CFTR gene: clinical and laboratory implications for cystic fibrosis screening. Hum Genet. 2006;119(1-2):126–136.
[hr]
Dr. Chen and Dr. Quan are senior directors, Department of Molecular Genetics, and Cathi Franklin is manager of the Genetic Counselor Organization—all at Quest Diagnostics, San Juan Capistrano, Calif.
[hr]
Test yourself
Here are three questions taken from the case report.
Answers are online now at www.amp.org/casereports and will be published next month in CAP TODAY.
1. Hb H disease is usually caused by the loss of expression of what number of alpha-globin genes?
a) one
b) two
c) three
d) four
2. Which of the following is not one of the seven common alpha-thalassemia deletion mutations?
a) alpha3.7 deletion
b) –SEA deletion
c) –FIL deletion
d) HS-40 deletion
3. Hemoglobin HPLC analysis may not be able to detect Hb H, due to its:
a) low molecular weight
b) poor stability
c) high molecular weight
d) low quantity
[hr]