
William Check, PhD
March 2018—From phenotype to genotype in the understanding and diagnosis of cardiovascular disease—that was the medical journey on which Joseph Maleszewski, MD, and Birgit Funke, PhD, took attendees at a symposium at the November 2017 meeting of the Association for Molecular Pathology.

Traditional techniques have paved the way to targeted molecular testing and individualized medicine in cardiovascular disease. “Specifically with regard to cardiovascular disease, there is a growing recognition that neither traditional phenotypic nor genotypic information in isolation is particularly useful in terms of understanding the entire clinical picture,” Dr. Maleszewski, a professor of laboratory medicine and pathology and of medicine at Mayo Clinic, Rochester, Minn., said in an interview. “We have become increasingly aware that both clinical and pathological phenotype as well as genotype need to be married up to make informed decisions about patients and family members.”
Many seem to believe that answers will be completely contained within the genome, he says. “Certainly for cardiovascular disease we know that won’t be the case.”
Dr. Funke spoke about the design and validation of genetic tests for inherited cardiomyopathies. “These are very prevalent disorders with a strong genetic basis and a potentially severe outcome. So genetic testing for both diagnostic and predictive purposes is important,” she tells CAP TODAY.
Genetic testing is initiated today when a person is symptomatic, and, combined with family history, it can release some family members from lifelong screening. Are we ready to move from reaction to prediction? asked Dr. Funke, VP of clinical affairs at the private startup Veritas Genetics and associate professor of pathology (part-time), Harvard Medical School. “It is not as easy as it seems.”
To understand the causes of sudden death, Dr. Maleszewski and colleagues have been examining autopsy reports in a group of college athletes who died suddenly. They reported: “Unexplained death with a structurally normal heart is the most common finding after suspected sudden cardiac death in NCAA athletes. Hypertrophic cardiomyopathy is infrequently seen, and conclusions in autopsy reports may not accurately reflect the pathological findings” (Harmon KG, et al. Circ Arrhythm Electrophysiol. 2014;7[2]:198–204). “Unfortunately,” Dr. Maleszewski says, “many of these cases are difficult to adjudicate because of the varied ways in which these cases are approached across the country. Resources for evaluating sudden death are certainly different from jurisdiction to jurisdiction.”
To Dr. Maleszewski, these observations support the need for widespread access to combined pathologic and genetic analysis. “It is important to start taking a closer look at cases where there is no clear finding at autopsy,” he says. For those cases, “we are advising that they seek out a subspecialist consult to look for subtle findings that may direct where to look in the genes for culprit lesions. Having genetic tests available makes a better pathologic exam more useful.”
Speaking to CAP TODAY, Dr. Maleszewski, who is section head of cardiovascular pathology at Mayo, described a recent case that highlights the importance of specialist-guided postmortem genetic testing. An apparently healthy teenage athlete died suddenly in her home. “We did a postmortem,” he said, which included the usual gross and microscopic exams. “Her heart was only a little bit heavier than we expected—slightly too heavy for a young woman of her size but within the spectrum of normal for an adult, which she could easily have been considered by some.”
The histologic findings were subtle, but also suspicious, raising concerns she might have had hypertrophic cardiomyopathy, and genetic testing corroborated the suspicion. “The most important genetic result was that the test was positive for a mutation known to be associated with that condition,” Dr. Maleszewski said. “We used that information to screen the decedent’s relatives, thereby ruling about half of them out of further testing and intervention and focusing screening on those at highest risk. In the old algorithm, we would have done ongoing rigorous physical exam of all surviving family members until they manifested disease.” Given the subtle nature of her phenotype, he said, “it is conceivable that the disease may not have been picked up as early in some of her family members without the positive genetics.”
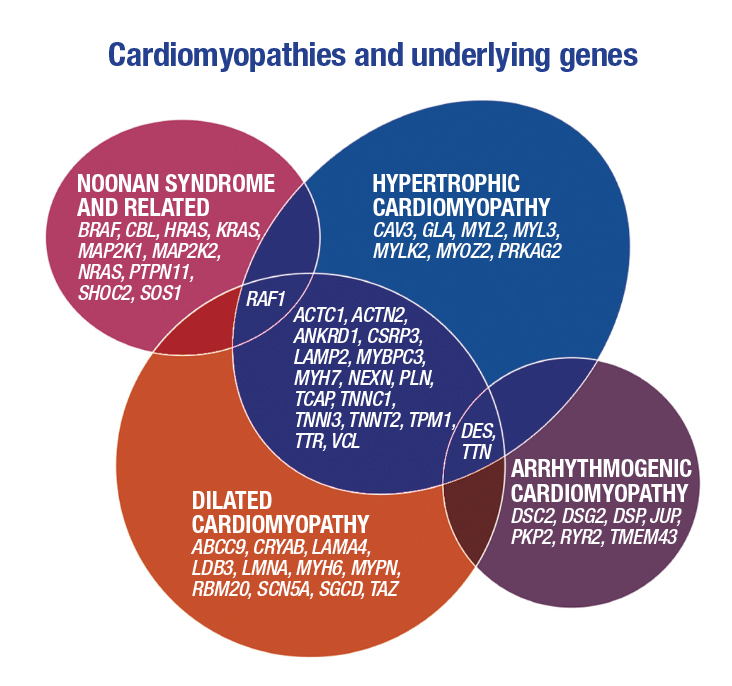
Dr. Maleszewski noted the three major types of cardiomyopathy—dilated, hypertrophic, and arrhythmogenic—and presented in Venn diagram format the many underlying genes (at right). The diagram illustrates the considerable overlap among these conditions from a genetic standpoint, especially between hypertrophic and dilated cardiomyopathies, the most common forms.
Molecular testing can be used to screen asymptomatic family members, follow kindreds, and provide prognostic information. While the presence or absence of a mutation is somewhat prognostic, individual mutations are not predictive of a patient’s course, he said.
Molecular testing, despite its potential utility, is not routinely used in cases of sudden death with negative autopsy results. Dr. Maleszewski cites three reasons. In autopsy-negative cases, a toxicological cause of death is the usual presumption, even though it is not explanatory in about 40 percent of cases. Blood is rarely collected for molecular genetic testing, and formalin-fixed, paraffin-embedded tissue, which is the most frequently stored material, is not optimal for DNA analysis. And molecular testing is expensive—$2,000 to $3,000.
In an interview, Dr. Maleszewski explained that FFPE tissue is often viewed as suboptimal for molecular analysis because formaldehyde cross-links DNA and shears it into small fragments that are difficult to sequence. He and colleagues have devised a protocol based on next-generation sequencing that overcomes this obstacle. “Two months ago we put out the first clinically validated molecular assay that allows us to interrogate FFPE samples,” he says. “It’s a game changer. Now we can go back and look at tissue that coroners and medical examiners have archived and test those samples to try to get more definitive answers for families.”
The assay was clinically validated on a panel of 101 CV-associated disease genes. He and colleagues reported last year that high-quality DNA was obtained from FFPE tissue samples as old as 15 years (Baudhuin LM, et al. Circ Cardiovasc Genet. 2017;10[6].doi:10.1161/circgenetics.117.001844). They found that 99.8 to 99.9 percent of bases had read depths greater than 100X. Most important, they write, “Genomic analysis of FFPET [FFPE tissue] from the 4 phenotype-positive/genotype unknown cases all revealed putative disease-causing variants.”
Mayo Clinic now offers inherited CVD panels based on this assay: one for heritable connective tissue disease (18 genes), another for heritable arrhythmia syndromes (20 genes), and a third for cardiomyopathies (58 genes).
Molecular genetic testing for inherited cardiomyopathies is relatively recent, Dr. Funke told symposium attendees. It was only in 2003 that testing became available for hypertrophic cardiomyopathy. In 2011 a pan-cardio panel that covered genes for all three types of cardiomyopathies was first offered (Maron BJ, et al. J Am Coll Cardiol. 2012;60[8]:705–715).
“We were the first lab to introduce that testing,” Dr. Funke told CAP TODAY, referring to the Partners HealthCare Laboratory for Molecular Medicine (LMM), where she was director of clinical research and development. Although the LMM continues to offer subpanels, most physicians order the comprehensive panel. “It can be difficult to distinguish cardiomyopathies on a clinical basis, especially arrhythmic and dilated cardiomyopathy,” Dr. Funke says. “When clinicians see patients with cardiac symptoms, they work toward a clinical diagnosis. Genetic testing can augment the clinical diagnosis by differentiating between seemingly similar clinical presentations.
“On the other hand, sometimes the diagnosis is very clear. So then the clinician orders a targeted subpanel.” If the pan-cardio panel is ordered, she says, the chance of getting an inconclusive result—a variant of uncertain significance—increases.
During the decade that molecular genetic testing for inherited cardiomyopathies has been offered, two important observations have been made: detection rates are moderate, and locus heterogeneity is the norm. In large surveys, a pathogenic variant has been found in 32 percent of hypertrophic cardiomyopathy (HCM) and 37 percent of dilated cardiomyopathy (DCM) patients. Two genes—MYBPC3 and MYH7—accounted for 80 percent of the HCM cases, with the remainder spread among many mutations. A similar situation obtains with DCM.
Allelic heterogeneity is also the rule: Among 3,000 HCM probands, 63 percent of variants have been seen only once—so-called private mutations.
Another finding is that the traditional genetic testing approach—one panel at a time—can lead to diagnostic odysseys due to clinical heterogeneity. Dr. Funke shared a case in which the clinical diagnosis was DCM. The DCM panel detected a variant of uncertain significance, which did not segregate with disease in the family. In a second clinical exam, the diagnosis was revised to arrhythmogenic right ventricular cardiomyopathy (ARVC). The ARVC panel yielded a likely pathogenic variant. About three percent of cases diagnosed as DCM carry a pathogenic variant in an ARVC gene. “Traditional step-by-step testing does not make sense for disorders with clinical overlap,” Dr. Funke said. “Multidisease testing facilitates diagnosis.”
Deciding which genes should be included on a cardiovascular disease gene panel requires establishing the core values of genomic medicine: analytical and clinical validity and clinical utility. Clinical utility—the risks and benefits resulting from test use—can be especially tricky. While adding more genes might detect more affected individuals, it introduces the risk of detecting more variants of uncertain significance.
Broad sequencing has dramatically increased the number of putative disease genes (reviewed by Walsh R, et al. Clin Chem. 2017;63[1]:116–128). “Diagnostic sequencing has the potential to become an integral part of the clinical management of patients with inherited cardiac conditions,” the authors wrote. They cautioned, “[E]xpanded testing requires great rigor in the identification of pathogenic variants, with domain-specific knowledge required for variant interpretation.”
Dr. Funke agrees. For many published gene-disease associations, she said, “Claims typically rest on identification of rare variants, usually with little additional supporting data.”
The variety of genes included on HCM panels offered by 45 laboratories surveyed two years ago illustrates the uncertainty about pathogenic genes. Most of the labs in a subset of 14 had 10 to 40 genes on their panels. One had 50; another had 90.
Dr. Funke and colleagues analyzed their data for 766 patients tested with DCM panels from 2007 to 2011. During that time the panel grew from five genes to 46. Clinical sensitivity increased from 10 percent to 37 percent. Inconclusive findings also grew, from about 10 percent to about 60 percent (Pugh TJ, et al. Genet Med. 2014;16[8]:601–608). The investigators noted that “[T]he contributions of individual genes and the pathogenic variant spectrum are still poorly defined.” And they concluded, “Our data illustrate the utility of broad gene panels for genetically and clinically heterogeneous diseases but also highlight challenges as molecular diagnostics moves toward genome-wide testing.”
A variant of uncertain significance finding can affect patients differently, Dr. Funke says. “Depending on how you’re wired, it can be a concern or not. You may assume you have a pathogenic variant or that it signifies nothing.”
Dr. Funke shared a recently reported case in which the finding of a variant of uncertain significance had a powerful adverse clinical impact (Ackerman MJ. Heart Rhythm. 2015;12[11]:2325–2331). The proband died suddenly. The proband’s sister saw a cardiologist but her evaluation was unremarkable. Pan-arrhythmia testing detected a variant of uncertain significance, which the cardiologist concluded must have been the cause of the proband’s death. A prophylactic implantable cardioverter-defibrillator was placed in the sister and in three of her children. Years later it was found that that variant does not affect protein function. When tissue from the proband was finally tested, it was negative.

“I am trying to stress that even a physician can misinterpret a VUS,” Dr. Funke says. “That VUS should not have led to the action the physician took.”
To avoid such problems, the Clinical Genome Resource, or ClinGen, is building and centralizing resources to define the clinical relevance of genes and variants (www.clinicalgenome.org). Three questions are crucial: Is this gene associated with a disease? Is this variant causative? Is it actionable?
The ClinGen hypertrophic cardiomyopathy gene curation team is looking at 55 selected genes. Of the first 30 genes curated, only nine had strong or definitive evidence for pathogenicity. Twenty had limited or no evidence. Only two fell in the ambiguous “moderate” category. “Now what I would like to see happen is that labs not even include those genes with limited or weak evidence on panels,” Dr. Funke says.
She is proud of what’s been accomplished over the years through this international effort: “This is what it takes: experts contributing time and expertise to meet a goal in the absence of adequate funding,” she says. Dr. Funke is co-chair of the ClinGen Cardiovascular Clinical Domain Working Group.
She shared another case, one that illustrates how a lack of standards for sequence interpretation can affect patients. A laboratory had interpreted a DCM patient’s variant as likely pathogenic. The patient came to the Laboratory for Molecular Medicine for a second analysis, where the variant was interpreted to be of uncertain significance. Dr. Funke called the other laboratory and resolved the discrepancy. “This is why we need concrete evidence-based standards on which to base our interpretation.”
Since that time, a working group of the American College of Medical Genetics and Genomics and Association for Molecular Pathology published standards and guidelines for interpreting sequence variants (Richards S, et al. Genet Med. 2015;17[5]:405–424). However, “The new criteria are not the final answer,” she said. Three experts from the ClinGen Inherited Cardiomyopathy Expert Panel classified 10 MYH7 variants twice. Using their institutional criteria, concordance was 95 percent. Using the ACMG/AMP criteria, it was only 30 percent. Part of this problem is that the experts were not yet familiar with the new criteria. Another reason: The ACMG/AMP criteria are not specific to cardiomyopathy. “We need to make the criteria tighter with regard to specific diseases,” Dr. Funke says.
To address this need, the ClinGen Cardiovascular Clinical Domain Working Group has adapted and validated the existing variant classification framework for MYH7-associated inherited cardiomyopathies. The recommendations of the expert panel were published online ahead of print on Jan. 4 (Kelly MA, et al. Genet Med. 2018. doi:10.1038/gim.2017.218).
Sharing of sequence data among laboratories is one good way to enhance validation. Much variant-disease association data are public. However, Dr. Funke explains, “Many diagnostic labs have internal data they accumulate over time”—so-called private data. More than 3,000 patients have been studied at her own institution. “From each patient you learn something. Is that variant pathogenic or not?” Unless each laboratory shares its information publicly, though, it all remains in separate silos. “On the other hand,” Dr. Funke said, “once we pool those data, the number of times a variant has been seen increases, which can help clarify its clinical significance. For example, we may cross a threshold where we can confidently move a variant from likely pathogenic to pathogenic.”
One exercise conducted among ClinGen Inherited Cardiomyopathy Expert Panel members illustrates this point. When private data were added to public data, 12 of 60 variants analyzed (48 percent) changed classification. Of those, seven were upgraded from likely pathogenic to pathogenic. Reclassification was accomplished by sharing among “a handful” of laboratories (among them some of the world’s largest cardiomyopathy testing laboratories). As part of their mandate from ClinGen, the NIH-funded working groups are sharing their data in ClinVar.
The Sarcomeric Human Cardiomyopathy Registry consortium, too, observed greater agreement in hypertrophic cardiomyopathy genetic test interpretation with data sharing. “Discordance in variant classification among hypertrophic cardiomyopathy centers is largely attributable to privately held data,” investigators concluded (Furqan A, et al. Circ Cardiovasc Genet. 2017;10[5].doi:10.1161/circgenetics.116.001700). Says Dr. Funke, “We were happy to see they reached the same conclusions we did.”
Dr. Funke presented several suggestions for how to achieve complete harmonization of variant classification. Among them: enhance data submissions, create structured fields for case-level clinical data in public variant databases, and standardize clinical data collection by testing laboratories.
Looking to the next step, Dr. Funke raised the possibility of genetic screening for cardiovascular disease in the general population. “Let’s move forward, but let’s do it carefully and thoughtfully,” she advises. “It is more complicated than people have thought, including myself. We’re finding out we don’t know enough.”
[hr]
William Check is a writer in Ft. Lauderdale, Fla.