 CAP TODAY Pathology/Laboratory Medicine/Laboratory Management
CAP TODAY Pathology/Laboratory Medicine/Laboratory Management

Fragile X is the most frequent form of inherited mental retardation; it segregates as an X-linked dominant. (It is called “fragile” X because the terminal telomere of the X chromosome looks like it’s falling off.) Its major phenotype is intellectual disability, with average IQ around 40, as well as considerable social anxiety. Physically, FXS has a phenotype that Dr. Warren called “distinctive but subtle.”
In 1991 FXS became the first triplet repeat disorder identified, when a team led by Dr. Warren identified expansion of CGG triplet repeats in the FMR1 gene as the basis of the syndrome (Verkerk AJ, et al. Cell. 1991;65:905–914; Kremer EJ, et al. Science. 1991;252:1711–1714; Pieretti M, et al. Cell. 1991;66:817–822). We all have a sequence of six to 54 CGG repeats in our FMR1 gene; when it expands to more than 200 repeats, the full mutation ensues. Between 55 and about 200 repeats is a territory called premutation.
Dr. Warren’s latest work challenged “the untested belief that all fragile X is due to repeat expansion,” he told the AMP audience. In a later interview he said: “Repeat expansion gives a null mutation, a loss of function. We know from standard genetics that you can get a null mutation through more conventional mutations.” And yet only one missense mutation in FMR1 had been reported in 20 years of testing.
Dr. Warren’s group used next-generation sequencing to search for point mutations in 963 under-18 males referred for clinical testing for CGG expansion who were found to have normal repeat length.
In an example of defensive medicine, most children in the U.S. who don’t meet developmental milestones or who have behavioral issues are tested for FMR1 repeat expansion. With such nonspecific criteria being used as an indication for CGG expansion testing, the positive diagnostic yield of this test in most laboratories is typically less than two percent, leaving most cases unexplained.
Dr. Warren’s group found many novel variants among these 963 patients, suggesting to the investigators that “FMR1 sequence variants may represent an important cause of developmental delay” (Collins SC, et al. Am J Med Genet A. 2010;152A:2512–2520). Further investigation in Dr. Warren’s laboratory identified seven confirmed potentially pathological variants. He presented results of the extensive workup of three of the variants.
One boy with a T→C variant at the highly conserved position 746 showed features not typical for FXS, except for an IQ of 47. He had stereotypic behavior, delayed developmental milestones, and a previous diagnosis of autism spectrum disorder and ADHD. In 1997 it was shown that FMRP is translated near synapses in response to neurotransmitter activation. Using an in vitro system, Dr. Warren’s group showed that the *746T→C mutation eliminated this activity-driven translation of FMRP. “We thought that local synthesis of FMRP in response to synaptic activation was important, but there was no true evidence,” Dr. Warren told CAP TODAY. “This experiment shows that not being able to synthesize FMRP in response to synaptic activity is actually detrimental.” Here we can see the reciprocal synergy of investigating FMR1 variants at the molecular and cellular level: It illuminates the basis of clinical pathology while at the same time elucidating basic synaptic function.
A second patient, with an R138Q variant, had an IQ of 42 and developmental delay but otherwise no typical FXS features. He also had intractable seizures. To Dr. Warren and colleagues, this looked like a case of maternal transmission because his mother, who also had the R138Q variant, had problems in school and severe social anxiety. “Typically, if you have a mutation in the X chromosome, it presents as a more mild phenotype in the female,” Dr. Warren says.
In vitro work showed that FMRP from this patient could not rescue a defect in presynaptic function (See “Synaptic transmission”), making it the first example of a presynaptic defect in a patient (without the FXS phenotype). “Pretty much the whole field thinks that FMRP acts in the post-synaptic space,” Dr. Warren says. “But these data show that it is both pre- and post-synaptic. The phenotype of this patient with a defect in FMRP is much broader than what we previously expected.”
A third patient, who had the typical facies of FXS along with global developmental delay, severe intellectual disability, and autism spectrum disorder/ADHD, had normal repeat length but a single-nucleotide variant at the highly conserved 266 position of FMR1. His brother had the same variant, but a borderline IQ, showing the phenotypic spectrum of variants in FMR1. In vitro work showed that FMRP from this patient is severely defective in function (see box) and that the amino acid change disrupts its structure. Dr. Warren concluded that this G266E variant is a functional null mutation, just as he had postulated. So a single-base variant can cause classic FXS without CGG expansion.
Synaptic transmission
When it comes to the central nervous system, the synapse is where the critical action takes place. And it’s here, gathering evidence suggests, that the FMR1 gene, through its protein product FMRP, exerts its effect.
In his AMP award lecture, Dr. Stephen Warren said that FMRP is a regulator of synaptic translation. It selectively binds about three percent of brain mRNAs, primarily synaptic messages, blocking their translation. He and others reported some time ago that in a majority of male fragile X patients, FMR1 mRNA is missing, implying an absence of FMRP (Pieretti M, et al. Cell. 1991;66:817–822). “Absence of FMRP results in constitutive translation of certain synaptic proteins and leads to a weaker synapse,” Dr. Warren told attendees at the AMP meeting last year. (See box at right.)
“It is known that regulation of translation of the activated synapse is important for learning and memory,” Dr. Warren said in an interview. These cognitive functions are exactly what is impaired in fragile X syndrome. “If regulation of translation is abnormal, then you get fragile X syndrome,” Dr. Warren says. It seems that even for protein synthesis you can have too much of a good thing.
Work by Dr. Warren’s group with the newly discovered single-nucleotide variants in FMR1 fits into this conceptual scheme and adds new details to it. For instance, in the patient with the G266E mutation, FMRP does not bind mRNA or associate with polysomes. It has completely lost the ability to regulate protein synthesis. So it is not surprising that the boy with this variant had the full FXS picture.
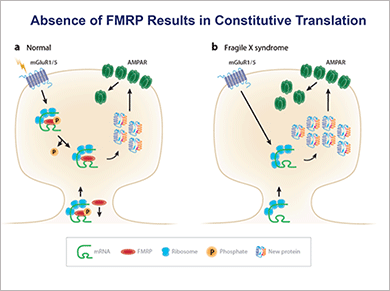
FMRP binds to mRNA in polysomes in synaptic neurons and modulates its translation. Absence of FMRP allows unregulated translation of mRNAs, producing excessive internalization of the AMPA receptor, a signal of a weakened synapse.
In contrast, FMRP from the patient with the R138Q variant binds mRNA and regulates translation. However, R138Q FMRP mutant FMRP does not rescue a presynaptic defect. A group led by Vitaly Klyachko, PhD, of Washington University, showed that knockout mice lacking FMRP have an abnormal action potential (Deng P-Y, et al. Neuron. 2013;77:696–711). Working in collaboration, the two labs showed that R138Q FMRP does not restore a normal action potential. “This is a whole new function that may be clinically relevant,” Dr. Warren says. “The action potential is pretty fundamental in neuronal transmission. Any abnormality is most likely deleterious.”
As Dr. Klyachko and his colleagues wrote in an earlier paper, “Activity-dependent presynaptic processes give rise to several forms of short-term plasticity, which is believed to control some essential neural functions, including information processing, working memory, and decision making” (Deng P-Y, et al. J Neurosci. 2011;31:10971–10982).
In vitro analysis of the *746T→C mutation revealed a different defect. It is known that FMRP is translated near synapses in response to neurotransmitter activation. Dr. Warren’s group showed that the *746T→C mutation eliminates this activity-driven translation of FMRP. Further investigation suggests how this happens. Messenger RNA was extracted from cells of healthy persons and from cells of the patient. When these mRNAs were mixed with brain lysate, normal mRNA bound a specific protein that mRNA from the patient did not. Dr. Warren’s group identified this protein as HuR, which is known to bind RNA and modulate translation.
Further work showed that HuR binds specifically to the sequence around the 746 locus of the FMR1 gene. As expected, purified HuR binds mRNA complementary to this region. However, it does not bind *746T→C mRNA. Taken together, this evidence delineates a system of protein synthesis regulation that is disrupted in the presence of the *746T→C mutation, creating yet another route to a faulty synapse.
—William Check, PhD
As understanding of the basis of FXS has advanced, therapeutic possibilities have arisen (see, for example: Bear ME, et al. Trends Neurosci. 2004;27:370–377; Henderson C, et al. Sci Transl Med. 2012;4:152ra128). Clinical trials have focused on two types of agents: drugs that antagonize metabotropic glutamate receptors (mGluR) and GABA agonists, which activate the gamma-aminobutyric acid receptor. The underlying rationale is that these agents may help restore regulatory function to FMRP-deficient neurons. Dr. Warren said that GABA agonists look hopeful but that one failed a trial because of the outcome criteria used. The drug’s sponsors chose an increase in cognitive function, which the drug didn’t accomplish. But the drug did improve socialization and anxiety. “Parents value those improvements,” he said, adding that other drugs “show hopeful signs.”

Regarding the three point mutations that Dr. Warren discussed, Dr. Lyon found the evidence that they caused the patients’ symptoms to be compelling. “The in vitro evidence gave me more confidence that these really are pathogenic,” she says. “It was much more convincing than the computed evidence that is often the only tool available to assess many novel variants.”
Dr. Schrijver says these mutations may contribute to these phenotypes. “However, due to the relative sparsity of data, we have the difficulty of making firm genotype-phenotype correlations. It will be important to see more mutations,” she says. “The more patients we can study the more comfortable we can be in interpreting the effects [of single-nucleotide variants] on such patients.”
Dr. Warren is continuing to look for such patients and his group is evaluating “a couple of others” who support its hypothesis.
More variants will be discovered, since Dr. Warren’s lab is offering exome sequencing for patients found to have a normal CGG repeat number in the standalone test. (Repeats are not picked up by sequencing.)
Dr. Schrijver thinks there is great potential for expanded testing in the future. “But it may be too early to implement something like this right now,” she says. “When you implement a clinical lab test it always lags behind what can be done in the research realm. You have to be sure that a properly validated test has clinical value and that you know how to interpret results.”
Dr. Warren acknowledges that “It will be some time before full exome sequencing is done on everyone.”
Dr. Grody views expanded testing favorably but with qualifications. “I was thinking to myself that we will have to change the way we diagnose fragile X and work up children with developmental delay,” he says. “However, first I would like to see more data. That’s why we go to meetings—to hear late-breaking information. But before we start sequencing every boy with developmental delay I would like to know more about yields and how often this happens.” Dr. Grody’s group does a lot of whole exome sequencing for many different indications, including developmental delay. “Over the past year we saw only one variant in the FMR1 gene, so perhaps this is not a very frequent finding.”
At the present time, Dr. Grody says, NGS is “terrible” at aligning repeat regions, so any boy suspected of FXS would have to have the standalone test for triplet repeats first. NGS will continue to get better, he says, adding, “There is no reason it couldn’t eventually pick up repeats.”
As for the cost-benefit, Dr. Hunt says it is a question of incremental pickup. “Will there be a good return on the additional investment?” she asks. “For many diseases you pick up the majority of cases with the most frequent mutations. Each time you add an uncommon mutation or alteration, you get a few more.” Testing can expand quickly, she notes, and good algorithms with which to test the cost-benefit ratio of each additional test are not available. “We will have to become more sophisticated to understand where we are going to draw limits. At some point we are gaining less and less. We need to figure out as a specialty and in health care in general where we draw limits, what’s valuable enough to add to the cost.”
Dr. Warren argues that the diagnostic yield of testing for point mutations in patients with normal CGG repeat lengths could approach one percent—his group found seven new variants in the 963 people tested. “With full exome sequencing we could see these all at once,” he says.
As for the added cost, “It is not really that much. These patients get tested for mutations in many genes, so adding another one isn’t really that costly,” he says. And not testing has a cost, too. FXS is not a lethal disease and requires lifelong care. “Societal costs are quite high,” Dr. Warren says.
Accumulating and analyzing additional single-nucleotide mutations in FMR1 should clarify how frequent these variants are and provide greater certainty about their pathogenicity, allowing optimal screening algorithms to be designed.
[hr]
William Check is a writer in Ft. Lauderdale, Fla.