
William Check, PhD
November 2014—When Birgit H. Funke, PhD, gave a talk earlier this year on incorporating bioinformatic tools and pipelines into medical NGS, at Molecular Medicine Tri-Con 2014, one of her slides showed the main bioinformatics activities needed to support sequencing. Among them were designing and building pipelines to manage genetic data, writing scripts for data analysis pipelines, and building custom applications.
But the point she emphasized most was “Clinical add-on: documentation + validation.” Dr. Funke, who is assistant professor of pathology at Massachusetts General Hospital/Harvard Medical School and director of clinical research and development for the Laboratory for Molecular Medicine at Partners HealthCare, said that working with an awareness of clinical application is new for bioinformaticians. In an interview with CAP TODAY, she illustrates this with a brief dialogue she had experienced:
Lab director: “Where did you store that script?”
Bioinformatician: “I don’t know, but I can rewrite it for you.”

While this attitude is all right for research work, it just won’t cut it in the world of CAP-accredited, CLIA-certified laboratory testing. “We have to validate everything, and that includes scripts,” Dr. Funke says. “Building a team of clinical bioinformaticians is a painful process. It is almost a new discipline.” It takes about a year, she estimates, for a bioinformatician to get on the same page with the rest of the laboratory.
“We need individuals who know how to code but who also understand genetics and the rigor of clinical lab work,” Dr. Funke adds. One big reason is that there is not much software designed for clinical next-generation sequencing. “I have yet to come across a lab that did not have to integrate those programs into their infrastructure,” she says. Bioinformaticians are equipped to do this. “It is critical to have at least one bioinformatician dedicated to your clinical analysis. There are more such people than before,” she says, “but they are still hard to come by.”
Why such an emphasis on trained bioinformaticians? Certainly in her Tri-Con talk, and in a webinar she gave in July in the “NGS 101 for the Clinic” series for the Association for Molecular Pathology, Dr. Funke described in detail the technical side of NGS and its associated informatics pipeline. But she said in the webinar: “The wet lab steps are the least of your problems. The more tricky issues surround bioinformatics analysis.” And it is for the latter that bioinformaticians are needed.

At Washington University School of Medicine, Rakesh Nagarajan, MD, PhD, and his colleagues also recognize that skilled, clinically aware bioinformaticians are essential for clinical NGS. “All of our bioinformaticians started as master’s-level personnel at the university’s Center for Biomedical Informatics,” which he directs, Dr. Nagarajan tells CAP TODAY. After a classical training in bioinformatics, they received training in NGS. They were then further trained in clinical concepts in the clinical lab and clinical IT environment. Traditional bioinformaticians are aware of pipelines and details and can tweak parameters in the research arena to see different outcomes, says Dr. Nagarajan, associate professor of pathology and immunology and of genetics. “In contrast, in the clinical arena we have one shot with patient data on a clinically validated pipeline, and that can’t be changed in an ad hoc fashion. We had to drill those concepts into them.”
There is a need, too, for pathologists with an understanding of bioinformatics pipelines. “There is a need for folks like myself who hacked away and trained ourselves,” Dr. Nagarajan says. “We kind of paved the road when there wasn’t one. In these times, we need interdisciplinary teams, including liaisons, who can speak across disciplines and can direct and train bioinformaticians and train clinicians and genomicists in the caveats of bioinformatics tools.” While he and his contemporaries essentially trained themselves, he says, “In the future there need to be formal training programs.”

A mastery of the techniques of clinical NGS, an appreciation of the complexities of an NGS bioinformatics pipeline, clinically oriented bioinformaticians—what more does a laboratory need to carry out accurate clinical NGS? Guidelines and standardization, says Ira M. Lubin, PhD, team lead for genetics in the Laboratory Research and Evaluation Branch at the Centers for Disease Control and Prevention. At the 2013 AMP meeting, Dr. Lubin organized a workshop featuring four members of a working group that he coordinates called Nex-StoCT (Next-generation Sequencing: Standardization of Clinical Testing). Drs. Funke and Nagarajan were two of the speakers in that workshop, titled “Upcoming Guidance for the Design and Optimization of a Clinical NGS Informatics Pipeline.”
The working group was a continuation of an effort that began in 2011, Dr. Lubin tells CAP TODAY. The first Nex-StoCT working group identified principles and developed recommendations for implementing and integrating NGS into clinical settings (Gargis AS, et al. Nat Biotechnol. 2012;30:1033–1036). “From that initial effort, there was high interest to convene a second working group on informatics design and optimization of a clinical informatics pipeline,” he says. That led to the ideas reported at the AMP session. A manuscript written by the second working group was submitted for publication recently.
The pace with which NGS was being adopted for use in the clinic, for inherited diseases and oncology, is what led to the formation of the working groups. In an interview, Dr. Lubin says the working groups have emphasized the limitation posed by the insufficient number of people with expertise to integrate NGS into practice. “The most important point made throughout all work groups was that if you are going to become involved in NGS and reporting results from your lab and plan to do analysis in your lab, you need to have informaticians familiar with the clinical specifications of NGS,” he says.
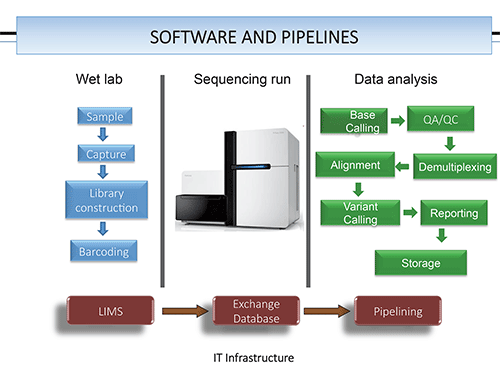
Clinical software is, for the most part, not available, he agrees. “We are still at the point where we are using software that was built and adapted from research settings.” Laboratories are running custom-designed informatics pipelines, he says, with the exception of two tests cleared by the FDA for cystic fibrosis. “Promoting the quality of NGS testing to produce reliable results” is the main goal of the guidelines coming from the working groups.
With the additional work required to translate sequencing from research to diagnostics, it’s not surprising that the much-touted $1,000 genome, while perhaps feasible in the near future for discovery, is not even on the radar for clinical work. Dr. Funke says the cost of sequencing a genome fell quickly until 2011, but has leveled off in the $8,000 to $9,000 range. “We have a ways to go till we reach the $1,000 clinical genome,” she adds.
Cost will affect reimbursement. Dr. Nagarajan says even with a theoretical $1,000 genome, “which we have seen in the research environment but not in the clinic,” obtaining reimbursement for whole-genome sequencing will be difficult.
In her webinar, which focused on technical and reporting issues, Dr. Funke said that NGS “has outpaced the usual time for adoption into the clinic.” She also noted a trend toward genomewide testing. “In the future, whole-genome sequencing will likely replace all of these techniques,” she predicted, referring to arrays, SNP chips, gene panels, and whole-exome sequencing. “That is still a few years in the future.”
NGS differs from the gold standard, Sanger sequencing, in several important ways. In Sanger, targets are amplified by PCR one exon at a time. In NGS, thousands of exons are amplified at one time. In Sanger, all molecules end up in the same tube for sequencing. In NGS, there is a physical separation of molecules during sequencing, leading to the term “massively parallel” sequencing.
Choosing the optimal hardware for your laboratory depends on turnaround time, volume, and target—the same parameters that govern the selection of other laboratory instruments. Tests that need a rapid TAT, for example, will require a smaller sequencer.
In its storage requirements, though, NGS differs radically from other tests, and from Sanger sequencing. With NGS, Dr. Funke said, “There is considerable cost associated with hardware. You can’t store that much data on a regular network anymore.” She showed that, coincident with the introduction of the first massively parallel sequencer in 2005, the 454 GS-20 pyrosequencer, the amount of data per run went from 10e2 to 10e14 (Mardis ER. Nature. 2011;470: 198–203). As a result, a laboratory doing NGS might need a high-capacity, high-performance storage system for data analysis (cost: approximately $1 million); 24 eight-core nodes with 2 Gb RAM per core; and a moderate-capacity, medium-performance storage system for permanent storage.
Huge data sets raise the question of which files to save. “It is unrealistic to save image files,” Dr. Funke said. “Fastq files and/or bam [binary alignment map] files are more realistic file formats, though those still take up a fair amount of storage space.” In the Partners Laboratory for Molecular Medicine, the bam file and the downstream filtered process file are saved along with the patient’s DNA. This is ultimately the most reliable storage medium, she says, because it allows re-running the assay with new technologies, if need be.
Increasing data outputs have another consequence. “When we started doing NGS in 2009, we only had two bioinformaticians,” Dr. Funke said. “Today we have tripled that number.”
Quality of sequencing is indicated by a number called the Phred score (named after Frederick Sanger). Sanger sequencing routinely achieves a Phred score of Q=40, for 99.99 percent accuracy, or one error per 10,000 base calls, she says. Most NGS has a Phred score of Q=30, indicating 99.9 percent base call accuracy, or one error out of every 1,000 bases.
NGS instruments still produce shorter read lengths than Sanger, which makes NGS alignment error prone and more computationally intensive, Dr. Funke said. “In 2011, you could get 50 base-paired end reads. Today, they are routinely 100 bp and above,” she says.
NGS will evolve to be the new gold standard, she believes, but for now many labs rely on Sanger for variant confirmation and filling in missing data. In particular, NGS has trouble with areas of severe sequence homology to other loci such as pseudogenes. Indels have high false-positive and false-negative rates with NGS. “It is still best to confirm variants found on NGS because of the inherent inaccuracy of this technology for some variant types,” Dr. Funke said.
Once variants are identified and collated in a variant caller format (vcf) file, it is necessary to apply filters of various kinds to remove commonly seen variants, which are not likely pathogenic. Other filters include one to ensure adequate coverage (read depth) and one to correct for strand bias (presence of the variant in one direction). Allelic fraction is also important. Dr. Funke cautions that an allelic fraction of 0.2, which works for substitutions (single nucleotide variants), may not detect all indels.
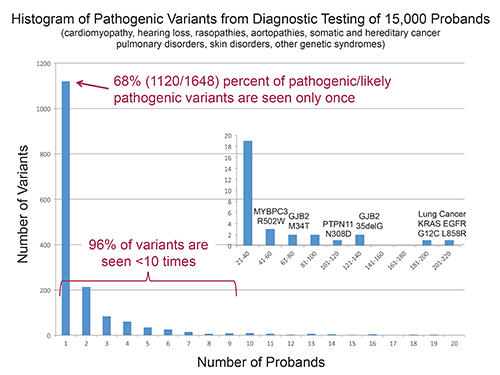
Time required to evaluate variants for pathogenicity has become the new bottleneck in NGS, Dr. Funke said, because the vast majority of variants seen in work with inherited disorders are novel. In diagnostic testing of 15,000 probands in the Laboratory for Molecular Medicine, 68 percent of 1,648 pathogenic or likely pathogenic variants were seen only once. (See “Histogram of pathogenic variants from diagnostic testing of 15,000 probands.”)
Gene sequencing has other bioinformatics challenges, such as the fact that the human reference sequence, which is used to interpret variants in a patient sample, contains pathogenic variants and risk alleles. “The human genome reference sequence to which we are all comparing our sequences contains real mutations,” Dr. Funke says. For instance, one version of the reference genome contains a variant in the gene for factor V Leiden, reflecting the genome of one person who gave reference DNA. “Imagine sequencing a person who is homozygous for that variant,” she says. “The variant caller thinks it’s normal when in fact this is medically significant.”
To deal with this complication, “we built a custom analysis tool that contains that knowledge and that tells the variant caller to go to certain sites we know are not wild type or are incorrect in the reference sequence and tells the machine to return the exact patient nucleotide at that position.” Interpretation is then done manually.
Considering the care and extra work needed to avoid errors in interpreting variants in human samples, it’s not surprising that the cost of clinical sequencing is higher than in research. “There is so much misconception about that,” Dr. Funke says. “When people quote the $1,000 genome, it stops after variants are called.” In clinical work, there is much more quality control, from sample accession and DNA extraction all the way to reporting the sequence. “Interpretation of variants doesn’t fall out automatically from the genetic data.” She cites the use of additional techniques, often Sanger sequencing, to verify variants and to fill in data that did not work in NGS. “We are striving for completeness and 100 percent accuracy, which is not necessary for research,” she says. “For someone like me, that means 30 minutes or more per case for a large NGS test such as a targeted 100-gene panel. And that’s after a lot of people before me have already done a lot of work.”
Does Dr. Funke think laboratories have an obligation to report variants found in sequencing that at the time of result reporting have no clinical implications? On disease-specific panels, her laboratory does report variants that do not have implications at the time of sequencing. “There is no one answer for all cases,” she says. “For our targeted panels, we report everything we find except for known benign variants. We do that because we can; the scope is limited.” Typically, one to three medically relevant variants occur per sample. “It is not possible to scale this to exome sequencing, where there may be 20,000 variants per person on average,” she says. “So we use a higher threshold and report likely pathogenic variants only.” A disclaimer in the report reads: “Please be aware knowledge can change. In the near term future, one of these variants may change significance.”
Speaking with CAP TODAY, Dr. Nagarajan addresses a broader question: whether to report everything found on clinical whole-exome and whole-genome sequencing, including incidental findings. Controversy has surrounded this question since the American College of Medical Genetics and Genomics announced recommendations in March 2013 (Green RC, et al. Genet Med. 2013;15:565–574). In its most important recommendation, the ACMG proposed that laboratories have a responsibility to seek and report mutations in a minimum list of 56 genes (23 of which are cancer related) regardless of test indication, patient preference, or age.
Dr. Nagarajan distinguishes two types of exome sequencing, saying: “We believe that the focus of those recommendations was around clinical assays claiming to sequence the entire exome. There is a slight nuance here. Places such as Baylor or Emory or UCLA are offering clinical exome sequencing for patients with developmental problems who are going through a diagnostic odyssey. They are looking at all genes and drilling down on those with certain phenotypes to simplify reporting. I believe that ACMG made the recommendations in that context.”
In contrast, he says, he and others are validating available whole-exome sequencing research products to report particular genes related to individual disease indications. The analytical component needs to be validated only once. “That allows you to determine the analytical specificity, and more importantly positive predictive value, and sensitivity. Once you have that, you can use the assay in several disease settings after confirming diagnostic specificity and sensitivity in each of those settings.” When sequencing a patient sample, all genes are captured, but only genes relevant to the patient’s disease condition are sequenced and analyzed. “That is negligibly different from doing a limited panel for the specific disease indication,” Dr. Nagarajan argues.
Like Dr. Nagarajan’s department, many other laboratories are moving to whole-exome sequencing. This will have an impact on the recommendations coming from the new CDC-facilitated work group that is addressing NGS sequence data standards, Dr. Lubin says. “There has been an evolution in our thinking since the AMP session,” he says. “At that time, most labs were focused on gene panels. Now many are moving to whole-exome sequencing, and eventually they will be performing whole-genome sequencing.
“With that movement,” Dr. Lubin says, “laboratories face the issue of generating much larger data sets that require more sophisticated analysis to generate results.” One major issue continues to resonate in the new work group: the recommendation that labs should align against a Genome Reference Consortium versioned reference assembly. A reference assembly consists of a standard genomic representation that includes a primary sequence plus alternative sequences, which are parts of the genome that can be mapped to a particular chromosome but fundamentally differ in sequence. Alignment against a versioned reference assembly as opposed to a set of discrete sequences promotes uniform assignment of variant positions and is useful to minimize forced alignment to a homologous region of the genome that can result in calling errors.
Gene panels don’t need a full assembly; they can be aligned to laboratory-selected sequences. Here, too, the work group raises a technical issue: developing standards for unambiguous representation of genomic sequences. “When you align to a sequence to describe a variant unambiguously, you not only say just what that variant is, but you also assign a set of genomic coordinates to describe where it resides on the reference assembly,” Dr. Lubin says. “The laboratory-selected sequence that you align to should be deposited to a recognized public database,” he says, such as those managed by the National Center for Biotechnology Information. “NCBI will map sequences deposited into their databases back to the reference assembly using uniform methods that will also generate genomic coordinates.”
OPEN Next-generation sequencers interactive product guide
The CDC-facilitated work group further recommends that the laboratory not take responsibility for aligning its laboratory-selected sequence against the reference assembly. “This is because aligners and their setup vary among laboratories, and this can result in different assignment of variant positions among laboratory settings,” Dr. Lubin says. In many instances this is not a problem, because the laboratory will use sequences derived from the NCBI RefSeq or comparable database, in which common methods have been used to cross-map positions back to the reference assembly.
Proficiency testing should be applied to NGS to provide a means to compare laboratory performance for those offering NGS testing. The CAP Next Generation Sequencing Project Team has developed a methods-based proficiency testing program for next-gen sequencing that will be available in early 2015.
Standards will also be necessary. In particular, Dr. Lubin emphasizes interoperability. “This will require standard protocols so that each lab is conforming to the same standard when communicating data through the health care system,” he says. If a laboratory generates a file with a set of variants, that file can be messaged or communicated to another laboratory that would have the technology to see and understand the contents of that variant file. The CDC-facilitated work group is working with the HL7 Clinical Genomics work group to address some of theses issues. The HL7 group is tasked with developing national and international standard recommendations for the messaging of genomic data through a health IT infrastructure.
“This is a bit into the future,” Dr. Lubin says, “but it is coming fast.”
[hr]
William Check is a writer in Ft. Lauderdale, Fla.
![]()