 CAP TODAY Pathology/Laboratory Medicine/Laboratory Management
CAP TODAY Pathology/Laboratory Medicine/Laboratory Management

Neng Chen, PhD, DABMGG
Cathi Rubin Franklin, MS, LCGC, DABMGG
Franklin Quan, PhD, DABMGG
![]()
February 2018—CAP TODAY and the Association for Molecular Pathology have teamed up to bring molecular case reports to CAP TODAY readers. AMP members write the reports using clinical cases from their own practices that show molecular testing’s important role in diagnosis, prognosis, and treatment. The following report comes from Quest Diagnostics. If you would like to submit a case report, please send an email to the AMP at amp@amp.org. For more information about the AMP and all previously published case reports, visit www.amp.org.
The alpha-globin gene locus (Fig. 1A) contains two alpha-globin genes: HBA1 and HBA2. An upstream regulatory region, known as HS-40, is essential for alpha-globin gene expression.1,2 The loss or inactivation of one or two alpha-globin genes is not clinically significant while the loss or inactivation of all four alpha-globin genes results in a lethal condition known as hemoglobin (Hb) Barts hydrops fetalis syndrome, the most severe form of alpha-thalassemia. The loss or inactivation of three alpha-globin genes results in a form of alpha-thalassemia known as hemoglobin H (Hb H) disease.
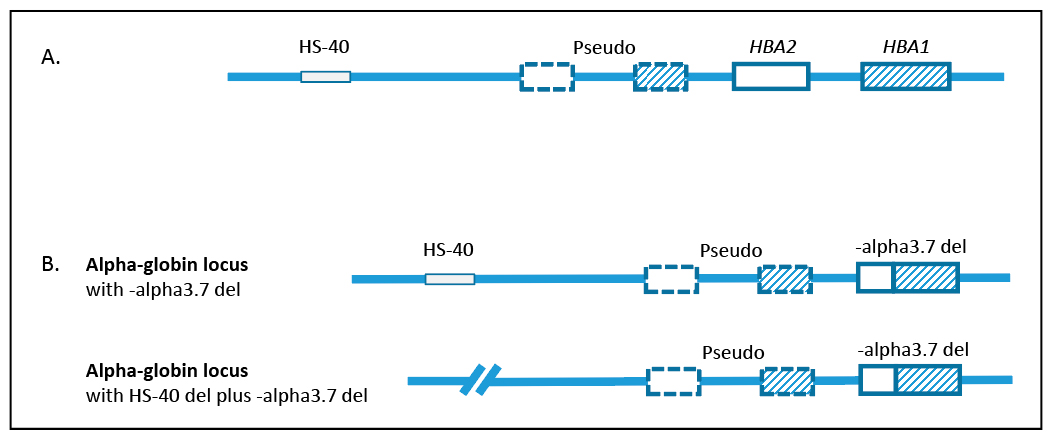
Fig. 1. Structure of the alpha-globin gene locus. A. Normal arrangement of HBA1, HBA2, and HS-40. Each alpha-globin gene locus contains two alpha-globin genes, HBA1 and HBA2. HS-40 is the upstream regulatory region required for alpha-globin gene expression. The alpha-globin gene locus also contains two non-functional pseudogenes. B. Genotype for the patient. One chromosome has the -alpha3.7 deletion; the other chromosome has the -alpha3.7 deletion and an HS-40 deletion.
Hb H disease has a variable phenotype. Hb H disease caused by deletions in the alpha-globin gene locus is associated with moderate anemia, microcytosis, and hypochromia. In contrast, nondeletional Hb H disease, caused by a point mutation or small insertion/deletion in one of the alpha-globin genes, is associated with a more severe anemia, and patients are likely to be transfusion dependent.
Patients with Hb H disease are often diagnosed after being seen for microcytic anemia or other indications, such as infection, hepatosplenomegaly, cholelithiasis, or severe anemia during pregnancy. For patients with low MCV, Heinz bodies after brilliant cresyl blue staining of blood smears, or Hb H and/or Hb Barts by high-performance liquid chromatography or gel electrophoresis, the differential diagnosis should include Hb H disease, hemolytic anemia, and alpha-thalassemia X-linked intellectual disability (ATRX) syndrome. Molecular genetic testing is used to confirm a diagnosis of Hb H disease and provide information for genetic counseling.
Patient case. A 50-year-old African-American male was evaluated by his primary care physician because of anemia. Several years prior, the patient had gastrointestinal bleeding and was diagnosed with colon cancer. He was anemic and transfused at that time as part of his treatment. His primary physician ordered blood smears and iron studies to investigate the cause of his anemia. The blood smears revealed microcytosis and the presence of target cells, indicating microcytic anemia. Iron studies indicated that all tested parameters were within the normal reference ranges (Table 1). Iron deficiency was ruled out as a cause of the patient’s anemia. The hemoccult test was positive at the time of his visit to the specialist.