 CAP TODAY Pathology/Laboratory Medicine/Laboratory Management
CAP TODAY Pathology/Laboratory Medicine/Laboratory Management

Shalini Verma, MD; Alessandra Ferrajoli, MD; Sa Wang, MD; Lynne V. Abruzzo, MD, PhD; Rachel L. Sargent, MD
![]() CAP TODAY and the Association for Molecular Pathology have teamed up to bring molecular case reports to CAP TODAY readers. Here, this month, is case No. 6. (See the February, August, and September 2013 and the May and June 2014 issues for the first five.) AMP members write the reports using clinical cases from their own practices that show molecular testing’s important role in diagnosis, prognosis, treatment, and more. Case report No. 6 comes from UT-MD Anderson Cancer Center in Houston. If you would like to submit a case report, please send email to the AMP at amp@amp.org. For more information about the AMP, visit www.amp.org.
CAP TODAY and the Association for Molecular Pathology have teamed up to bring molecular case reports to CAP TODAY readers. Here, this month, is case No. 6. (See the February, August, and September 2013 and the May and June 2014 issues for the first five.) AMP members write the reports using clinical cases from their own practices that show molecular testing’s important role in diagnosis, prognosis, treatment, and more. Case report No. 6 comes from UT-MD Anderson Cancer Center in Houston. If you would like to submit a case report, please send email to the AMP at amp@amp.org. For more information about the AMP, visit www.amp.org.
Myeloproliferative neoplasms and myelodysplastic/myeloproliferative neoplasms are clonal hematopoietic neoplasms that often share morphologic features. We report a rare case of two concurrent myeloid neoplasms: chronic myelogenous leukemia, BCR-ABL1 positive, chronic phase, and chronic myelomonocytic leukemia. This case illustrates the essential role of advanced molecular testing to achieve an appropriate diagnosis, prognosis, and therapeutic course.
Introduction
The 2008 World Health Organization classification of myeloid neoplasms includes myeloproliferative neoplasms (MPN); myelodysplastic syndromes (MDS); myelodysplastic/myeloproliferative neoplasms (MDS/MPN); myeloid and lymphoid neoplasms with eosinophilia and abnormalities of PDGFRA, PDGFRB, or FGFR1; and acute myeloid leukemias. The MPN are characterized by a clonal proliferation of one or more of the myeloid lineages and specifically include chronic myelogenous leukemia, BCR-ABL1 positive (CML); chronic neutrophilic leukemia; polycythemia vera; essential thrombocythemia; primary myelofibrosis; chronic eosinophilic leukemia; mastocytosis; and MPN, unclassifiable. MDS/MPN show myeloproliferative and dysplastic features and specifically include chronic myelomonocytic leukemia (CMML); juvenile myelomonocytic leukemia; atypical chronic myeloid leukemia, BCR-ABL1 negative; and MDS/MPN, unclassifiable. Evaluating the genetic aberrations outlined in the 2008 WHO classification for each of these distinct entities, using both cytogenetic and molecular techniques, is essential for precise classification, prognosis, treatment, and/or minimal residual disease monitoring.
Abbreviations: fluorescence in situ hybridization, FISH; hemoglobin, Hgb; hematocrit, Hct; mean corpuscular volume, MCV; white blood cell count, WBC; platelets, PLT.
Patient case
A 74-year-old woman presented in 2008 with anemia, thrombocytopenia, and leukocytosis with absolute monocytosis. Her medical and surgical history included hysterectomy in 1979; modified radical mastectomy, chemotherapy, radiation therapy, tamoxifen, and letrozole for estrogen and progesterone-positive breast carcinoma diagnosed in 1997; partial colectomy for diverticulitis in 2007; hypertension; hypothyroidism; and hyperlipidemia. A complete blood count and bone marrow examination performed at an outside hospital showed a myeloproliferative/myelodysplastic neoplasm best classified as chronic myelomonocytic leukemia (Table 1). The patient received interval observational management only.
In October 2011, the patient developed progressive leukocytosis, anemia, and thrombocytopenia. A CBC and bone marrow examination showed a hypercellular marrow (90 percent) with atypical megakaryocytic hyperplasia and dysgranulopoiesis. Conventional cytogenetic studies showed a complex karyotype including t(9;22). FISH studies showed a BCR-ABL1 rearrangement. Molecular studies for the JAK2 V617F mutation were negative (Table 1). The patient was diagnosed with chronic myelogenous leukemia, BCR-ABL1 positive. She was started on imatinib. Although she showed significant leukoreduction, she did not have a complete hematologic response, and she continued to have persistent anemia, thrombocytopenia, and monocytosis.
![Fig. A. Conventional cytogenetic studies show a complex karyotype: 47,XX,der(9)t(9;22)(q34;q11.2), ider(22)(q10)t(9;22),+ider(22)(q10)t(9;22)[3]/46,XX[17].](https://www.captodayonline.com/wordpress/wp-content/uploads/2014/11/Verma-et-al-Fig-A-300x222.jpg)
Fig. A. Conventional cytogenetic studies show a complex karyotype: 47,XX,der(9)t(9;22)(q34;q11.2), ider(22)(q10)t(9;22),+ider(22)(q10)t(9;22)[3]/46,XX[17].
In April 2012, the patient was referred to our institution for further evaluation and management. Physical examination showed no lymphadenopathy or hepatosplenomegaly. A CBC and bone marrow examination showed a hypercellular marrow with nine percent blasts, atypical megakaryocytic hyperplasia, mild dyserythropoiesis, monocytosis, and mild to moderate reticulin fibrosis. Conventional cytogenetic studies showed a complex karyotype including t(9;22) (Fig. A). FISH studies showed a BCR-ABL1 rearrangement and extra copies of the BCR-ABL1 fusion signal (Fig. B and Table 1).
Despite the cytogenetic findings, a BCR-ABL1 fusion transcript was not detected by real-time polymerase chain reaction. Nested PCR studies for the BCR-ABL1 transcript revealed an alternative 166-bp BCR-ABL1 transcript (Fig. C), which Sanger sequencing demonstrated to be a B2A3 BCR-ABL1 fusion transcript with partial deletion of exon 3 of ABL1 at the fusion junction. A mutation in codon 12 of NRAS (NM_002524: c.34G>Cp.G12R) was also identified (see Fig. D, page 56). Other molecular testing and sequencing results are shown in Table 1.
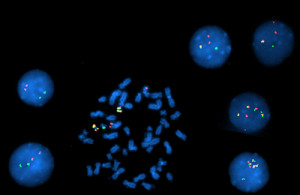
Fig. B. Interphase and metaphase FISH studies showed: A nuclear fusion signal with an extra red signal in 1.5 percent of the interphases indicating Ph+ cells; two nuclear fusion signals in two percent of interphases indicating Ph+ cells with an extra copy of the BCR-ABL1 gene fusion; four nuclear fusion signals in eight percent of interphases indicating Ph+ cells with three extra copies of the BCR-ABL1 gene fusion; 88.5 percent of the cells were Ph negative.
Given the overall clinicopathologic history, the differential diagnosis using the 2008 WHO classification was: 1) myeloproliferative/myelodysplastic neoplasm, unclassifiable; 2) therapy-related myeloproliferative/myelodysplastic neoplasm with acquisition of the Philadelphia (Ph) chromosome as a clonal evolution event; and 3) CML and concurrent CMML.
Results of conventional cytogenetic, FISH, and BCR-ABL1 molecular studies were integral in establishing an unequivocal diagnosis of CML, accelerated phase. Furthermore, this case shows many clinicopathologic features that are unusual for CML but support a diagnosis of CMML, including persistent monocytosis, granulocytic dysplasia, and the presence of an NRAS mutation. Hence, the patient was considered to have two separate diseases, CML and CMML, and was started on nilotinib and decitabine. After five months, nilotinib was discontinued owing to pancytopenia. Follow-up peripheral blood, bone marrow, cytogenetic, and molecular findings from February 2013 are shown in Table 1. To date, the patient has been receiving maintenance therapy with decitabine and is being monitored for persistent anemia and thrombocytopenia.
Discussion
Myeloproliferative neoplasms are characterized by the clonal proliferation of one or more of the myeloid lineages and typically occur in patients in the fifth to seventh decades of life. MDS/MPN include clonal myeloid neoplasms that pre-sent with morphologic findings consistent with MPN as well as dysplastic features that support a diagnosis of MDS. There is frequent morphologic overlap between these distinct neoplasms. Thus, a comprehensive clinical, morphologic, and molecular genetic evaluation should always be performed to facilitate accurate classification, appropriate treatment, and subsequent monitoring of minimal residual disease.
CMML is a clonal hematopoietic neoplasm with both myeloproliferative and myelodysplastic features. It is characterized by 1) persistent monocytosis (>1×109/L) in the peripheral blood; 2) absence of a Ph chromosome and BCR-ABL1 fusion gene; 3) no rearrangement of PDGFRA or PDGFRB; 4) fewer than 20 percent blasts in the peripheral blood and bone marrow; and 5) dysplasia involving one or more myeloid lineages. CMML can be diagnosed in the absence of convincing myelodysplasia if all other requirements are met and an acquired, clonal cytogenetic, or molecular genetic abnormality is present. A CMML diagnosis can also be made if monocytosis persists for at least three months and all other causes of monocytosis have been excluded. Although clonal cytogenetic abnormalities are found in 20 percent to 40 percent of patients with CMML, none are specific or diagnostic. Up to 40 percent of cases exhibit RAS point mutations at diagnosis or during the disease course. Thus, molecular testing for ABL1, PDGFRA, PDGFRB, and RAS mutations is important.