
CAP TODAY and the Association for Molecular Pathology have teamed up to bring molecular case reports to CAP TODAY readers. AMP members write the reports using clinical cases from their own practices that show molecular testing’s important role in diagnosis, prognosis, and treatment. The following report comes from the University of Pennsylvania Perelman School of Medicine. If you would like to submit a case report, please send an email to the AMP at amp@amp.org. For more information about the AMP and all previously published case reports, visit www.amp.org.
Guang Yang, MD, PhD
Gabriel C. Caponetti, MD
Jacquelyn J. Roth, PhD
Kojo S. Elenitoba-Johnson, MD
Megan S. Lim, MD, PhD
November 2020—Pediatric-type follicular lymphoma (PTFL) is a rare form of lymphoma that was recognized as a new diagnostic entity in the revised 2016 WHO Classification of Tumours of Haematopoietic and Lymphoid Tissues. The classic features of PTFL include male predominance, localized stage I lymphadenopathy, blastoid morphology, high proliferation index, and exceedingly good response rate to local excision.1 Because PTFL patients have an excellent prognosis, accurate and reliable identification of PTFL is important to minimize the prospect of unnecessary treatment.
Here we report an uncommon PTFL case that was diagnosed initially as “follicular lymphoma, WHO grade 1–2 of 3, with follicular growth pattern.” Using a massively parallel (next-generation) lymphoma sequencing panel, we were able to identify two missense variants in MAP2K1 and TNFRSF14. Although these variants are not unique to PTFL and have been reported in rare cases of other B-cell lymphomas, they provide an objective parameter to support the final diagnosis of PTFL.2
Case presentation. A 22-year-old male with no significant past medical history presented with a two-month history of painless swelling of the left neck. Physical examination showed a localized, rubbery, and non-tender left cervical nodule. CT imaging studies demonstrated a 1.8 × 1.6 × 2.4 cm solid mass in the upper left neck superficial to the sternocleidomastoid muscle. A PET/CT detected two hypermetabolic left cervical lymph nodes. An ultrasound-guided fine-needle aspiration biopsy of the mass showed an increased proportion of medium-sized to large lymphocytes in a background of small lymphocytes, and flow cytometry demonstrated a CD10+ CD5− mature B cell neoplasm. The patient subsequently underwent an excisional left cervical lymph node biopsy. Gross examination revealed a 3.5 × 1.7 × 0.6 cm tan-white soft tissue fragment, which was consistent with a lymph node with tan-pink cut surfaces.
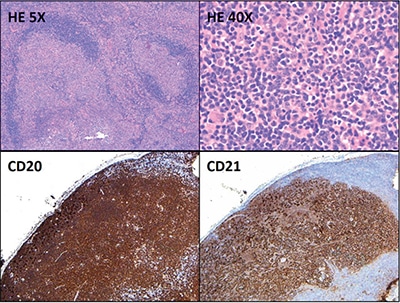
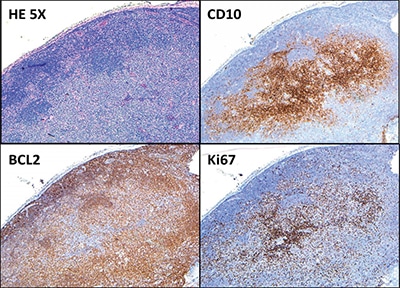
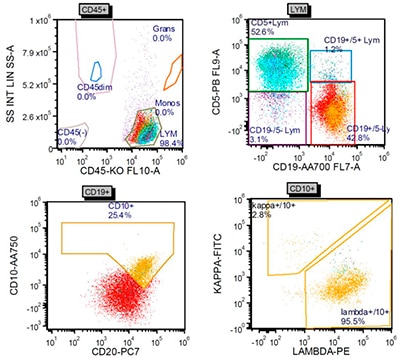
Hematoxylin-and-eosin–stained sections showed profiles of a lymph node with subtotal effacement of the architecture by a nodular infiltrate composed predominantly of small to medium-sized lymphoid cells with ovoid to irregular nuclei, clumped chromatin, inconspicuous nucleoli, scant cytoplasm, and an overall centrocytic morphology. Larger lymphoid cells morphologically compatible with centroblasts accounted for <15/high-power field within the nodules. No diffuse areas, sheets of large cells, or necrosis were seen. Immunohistochemistry showed that the majority of the cells within the nodules were positive for CD20, PAX5, CD10, BCL6 (partial), and BCL2 (partial, weak) (Figs. 1 and 2). CD21 and CD23 highlighted expanded and disrupted follicular dendritic cell meshworks. The proliferative index (Ki-67) was 20 to 30 percent within the nodules and 10 to 20 percent in the internodular areas. CD3 and CD5 highlighted an abundant number of small background T cells. Flow cytometry performed on a portion of the lymph node identified a surface lambda-restricted CD19+, CD20+, CD10+, and CD5– mature B cell population (11 percent of total events) (Fig. 3).
Fluorescence in situ hybridization was negative for BCL2 and BCL6 rearrangements and 1p36 deletion. A lymphoma sequencing panel (Illumina TruSight Sequencing Panel), designed and validated at the Center for Personalized Diagnostics at the University of Pennsylvania, was used for sequence analysis of 40 genes commonly altered in lymphomas (ATM, B2M, BIRC3, BRAF, BTK, CARD11, CD79A, CD79B, CIITA, CREBBP, CXCR4, EGR2, EZH2, GNA13, ID3, IDH2, JAK3, KLF2, MAP2K1, MYD88, NFKBIE, NOTCH1, NOTCH2, PLCG1, PLCG2, POT1, RHOA, RPS15, RRAGC, SF3B1, SOCS1, STAT3, STAT5B, TCF3, TET2, TNFAIP3, TNFRSF14, TP53, TRAF3, XPO1). Two missense variants were identified: MAP2K1 (NM_002755.3) c.308T>A, p.I103N (16 percent variant allele frequency) and TNFRSF14 (NM_003820.3) c.362G>A, p.C121Y (14 percent variant allele frequency) (Fig. 4). No variants were detected in any of the other 38 genes included in this panel.
Discussion. Recognized as a distinct diagnostic entity in the revised 2016 WHO classification, PTFL is a localized germinal-center–derived B cell neoplasm and has clinical, histopathologic, and genetic features distinct from typical follicular lymphoma. PTFL mainly affects young males and its prognosis is excellent. Studies have shown that local excision of PTFL diagnosed in children may be sufficient treatment, with no evidence of subsequent progression or relapse.1 Therefore, it is important to distinguish PTFL from typical follicular lymphoma to avoid unnecessary treatment.
Morphologically, the architecture of the involved lymph nodes is totally or subtotally effaced by large expansile follicles, often with a serpiginous growth pattern. The neoplastic follicles usually show a starry-sky pattern and thin or absent mantle zones.3 On high-power magnification, PTFL follicles consist of a monotonous population of intermediate-sized blastoid cells with frequent mitotic figures and inconspicuous nucleoli. Neoplastic cells in PTFL are usually positive for germinal center markers BCL6 and CD10, and negative or weakly positive for BCL2. The proliferative index (Ki-67) is usually high (greather than 30 percent). There is significant histologic and immunophenotypic overlap between PTFL and typical follicular lymphoma, sometimes making it difficult to differentiate between these two entities. In such situations, genetic studies may be helpful.
At the genetic level, in contrast to typical follicular lymphoma, PTFL lacks rearrangements of BCL2, BCL6, IRF4, or IGH, or amplification of BCL2, and has a near absence of mutations in epigenetic modifiers. The two most frequently mutated genes in PTFL are MAP2K1 and TNFRSF14 (observed in 43 percent and 29 percent of PTFL, respectively), whereas variants in epigenetic modifiers commonly found in follicular lymphoma (e.g. KMT2D, CREBBP, EZH2) are rare.1,4 In our case, no variants were identified in epigenetic modifiers such as EZH2 and CREBBP, which are commonly mutated in follicular lymphoma.1
Located on chromosome 1p36.32, TNFRSF14 encodes a member of the TNF (tumor necrosis factor) receptor superfamily. The encoded protein functions in signal transduction pathways that activate inflammatory and inhibitory T cell immune response. As one of the most commonly mutated genes in PTFL, TNFRSF14 variants are also frequently seen in typical follicular lymphoma (www.genecards.org).5 The observed TNFRSF14 variant (c.362G>A, p.C121Y) in our case has been described previously in other typical follicular lymphoma patients.6
MAP2K1 is located on chromosome 15q22.31 and its encoded protein is a member of the dual specificity protein kinase family, which acts as a mitogen-activated protein (MAP) kinase. MAP kinases, also known as extracellular signal-regulated kinases (ERKs), act as an integration point for multiple biochemical signals. As an essential component of MAP kinase signal transduction pathway, this kinase is involved in many cellular processes, such as proliferation, differentiation, and transcription regulation and development (www.genecards.org).5 MAP2K1 variants have been described as driver mutations in hairy cell leukemia variant, conventional hairy cell leukemia, Langerhans cell histiocytosis, and less frequently chronic lymphocytic leukemia, splenic marginal zone lymphoma, and splenic diffuse red pulp lymphoma.7 However, MAP2K1 alterations, to the best of our knowledge, have not been evaluated previously in typical follicular lymphoma cases.
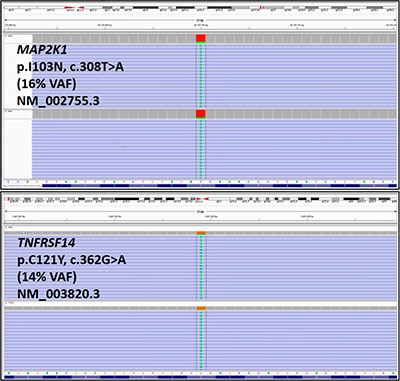
The MAP2K1 variant identified in the index case (p.I103N, c.308T>A) lies within the protein kinase domain of the MAP2K1 protein and is predicted to confer a gain of function as demonstrated by increased MAP2K1 auto-phosphorylation in cell culture.8 This MAP2K1 variant has been reported previously in classical hairy cell leukemia.9,10 In addition, a MAP2K1 variant on the same codon (103) has been detected in one other case of PTFL.7 Based on these lines of evidence, this variant is likely disease-associated in the setting of this patient’s PTFL.
Conclusion. The absence of BCL2 and BCL6 rearrangements and variants in epigenetic modifiers, the identification of MAP2K1 and TNFRSF14 mutations, and the clinical presentation (young male with cervical lymphadenopathy) all contributed in this case to the refinement of the initial diagnosis of typical follicular lymphoma to PTFL. This report emphasizes the importance of gene sequencing studies in the diagnostic workup of clinically challenging lymphoma cases.
- Swerdlow SH, Campo E, Harris NL, et al., eds. WHO Classification of Tumours of Haematopoietic and Lymphoid Tissues. IARC Press; 2017.
- Shamzah A, Fitzgibbon J. Pediatric-type FL: simply different. Blood. 2016;128(8):1030–1031.
- Jaffe ES, Arber DA, Campo E, Quintanilla-Martinez L, Orazi A, eds. Hematopathology. 2nd ed. Elsevier; 2017.
- Louissaint A Jr, Schafernak KT, Geyer JT, et al. Pediatric-type nodal follicular lymphoma: a biologically distinct lymphoma with frequent MAPK pathway mutations. Blood. 2016;128(8):1093–1100.
- Stelzer G, Rosen N, Plaschkes I, et al. The GeneCards suite: from gene data mining to disease genome sequence analyses. Curr Protoc Bioinformatics. 2016;54(1):1.30.1–1.30.33.
- Cheung KJ, Johnson NA, Affleck JG, et al. Acquired TNFRSF14 mutations in follicular lymphoma are associated with worse prognosis. Cancer Res. 2010;70(22):9166–9174.
- Schmidt J, Ramis-Zaldivar JE, Nadeu F, et al. Mutations of MAP2K1 are frequent in pediatric-type follicular lymphoma and result in ERK pathway activation. Blood. 2017;130(3):323–327.
- Kinoshita-Kikuta E, Kinoshita E, Ueda S, et al. Increase in constitutively active MEK1 species by introduction of MEK1 mutations identified in cancers. Biochim Biophys Acta Proteins Proteom. 2019;1867(1):62–70.
- Waterfall JJ, Arons E, Walker RL, et al. High prevalence of MAP2K1 mutations in variant and IGHV4-34-expressing hairy-cell leukemias. Nat Genet. 2014;46(1):8–10.
- Maitre E, Bertrand P, Maingonnat C, et al. New generation sequencing of targeted genes in the classical and the variant form of hairy cell leukemia highlights mutations in epigenetic regulation genes. Oncotarget. 2018;9(48):28866–28876.
- Robinson JT, Thorvaldsdóttir H, Wenger AM, Zehir A, Mesirov JP. Variant review with the Integrative Genomics Viewer. Cancer Res. 2017;77(21):e31–e34.
Dr. Yang was a hematopathology fellow (at the time the case report was written), Dr. Caponetti is assistant professor of clinical pathology and laboratory medicine, Dr. Roth is assistant professor of clinical pathology and laboratory medicine, Dr. Elenitoba-Johnson is the Peter C. Nowell, MD, professor and director of the Center for Personalized Diagnostics and of the Division of Precision and Computational Diagnostics, and Dr. Lim is professor of pathology and laboratory medicine and director of hematopathology—all at the University of Pennsylvania Perelman School of Medicine and the Hospital of the University of Pennsylvania. Dr. Yang is now an instructor, Department of Laboratory Medicine and Pathology, University of Minnesota Medical School.
Test yourself
[tabs type=”horizontal”][tabs_head][tab_title]Here are three questions taken from the case report. [/tab_title][tab_title]Answers[/tab_title][/tabs_head][tab]
1. Which statement is true about pediatric-type follicular lymphoma?
a. Its treatment usually involves only local excision.
b. Its prognosis is usually poor.
c. It usually harbors BCL2 and BCL6 rearrangements.
d. It mainly affects females.
2. Which of the following hematopoietic neoplasms commonly have MAP2K1 variants as driver mutations?
a. Hairy cell leukemia variant.
b. Langerhans cell histiocytosis.
c. Hairy cell leukemia.
d. All of the above.
3. Which statement is false about TNFRSF14?
a. It is one of the most common variants observed in PTFL.
b. Its encoded protein is a member of the TNF (tumor necrosis factor) receptor superfamily.
c. Its variants are specific for PTFL.
d. Its encoded protein activates the inflammatory and inhibitory T cell immune response.
[/tab]
[tab]
1. Which statement is true about pediatric-type follicular lymphoma?
a. Its treatment usually involves only local excision.
b. Its prognosis is usually poor.
c. It usually harbors BCL2 and BCL6 rearrangements.
d. It mainly affects females.
2. Which of the following hematopoietic neoplasms commonly have MAP2K1 variants as driver mutations?
a. Hairy cell leukemia variant.
b. Langerhans cell histiocytosis.
c. Hairy cell leukemia.
d. All of the above.
3. Which statement is false about TNFRSF14?
a. It is one of the most common variants observed in PTFL.
b. Its encoded protein is a member of the TNF (tumor necrosis factor) receptor superfamily.
c. Its variants are specific for PTFL.
d. Its encoded protein activates the inflammatory and inhibitory T cell immune response.
[/tab]
[/tabs]