
CAP TODAY and the Association for Molecular Pathology have teamed up to bring molecular case reports to CAP TODAY readers. AMP members write the reports using clinical cases from their own practices that show molecular testing’s important role in diagnosis, prognosis, and treatment. The following report comes from Washington University School of Medicine in St. Louis. If you would like to submit a case report, please send an email to the AMP at amp@amp.org. For more information about the AMP and all previously published case reports, visit www.amp.org.
Zita Hubler, MD, PhD; Catherine Gooch, MD
Erin Hediger, CGC; Yang Cao, PhD
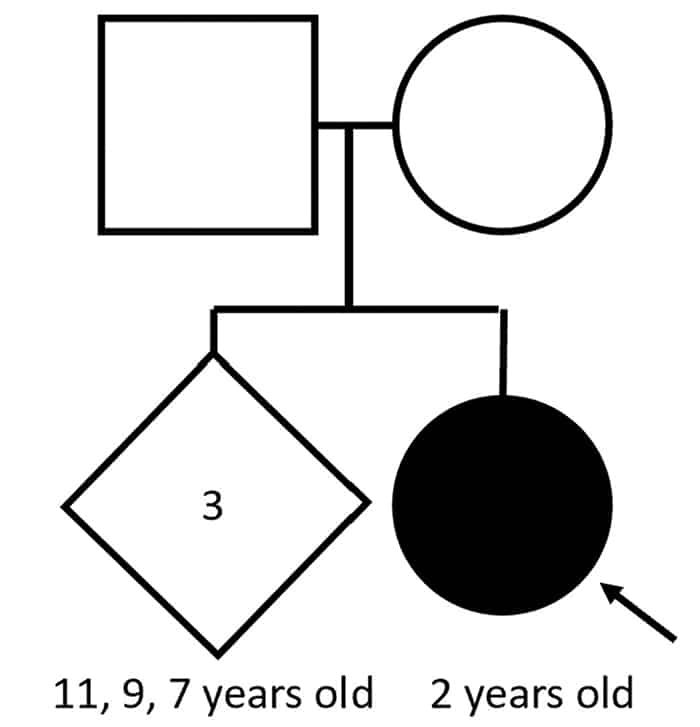
Case. A two-year-old female presented for a comprehensive plastic surgery evaluation for the repair of her cleft palate; this evaluation included a referral to medical genetics. The patient was born at full term to a 32-year-old woman with six total gestations and four live births. The pregnancy was uncomplicated, and the patient had an appropriate length, weight, and head circumference. A fully cleft palate was noted 24 hours after birth. Subsequently she missed several developmental, behavioral, and social functioning milestones. The family was concerned that she might have autism; therefore, a Modified Checklist for Autism in Toddlers (M-CHAT) screen was performed, on which she scored a six (medium/borderline risk for autism). The patient takes no medication but receives occupational and speech-language therapy. She lives with her family, including three full siblings. There is no family history of cleft palate or autism (Fig. 1A). A review of systems was notable for dental problems, drooling, and behavioral problems (agitation, self-injury). On the physical exam, the patient has an underbite, full cheeks, and prominent veins on the eyelids and bridge of the nose. She is nonverbal, aside from the word “no.” Her feet are notable for long toes and left greater than right fourth toe clinodactyly.
About 10 percent of individuals with cleft lip and palate have a chromosome condition that can be diagnosed with a chromosomal microarray. Further, chromosomal microarray is the first-line diagnostic test for patients with intellectual disability/developmental delay and congenital anomalies per American College of Medical Genetics and Genomics recommendations.1
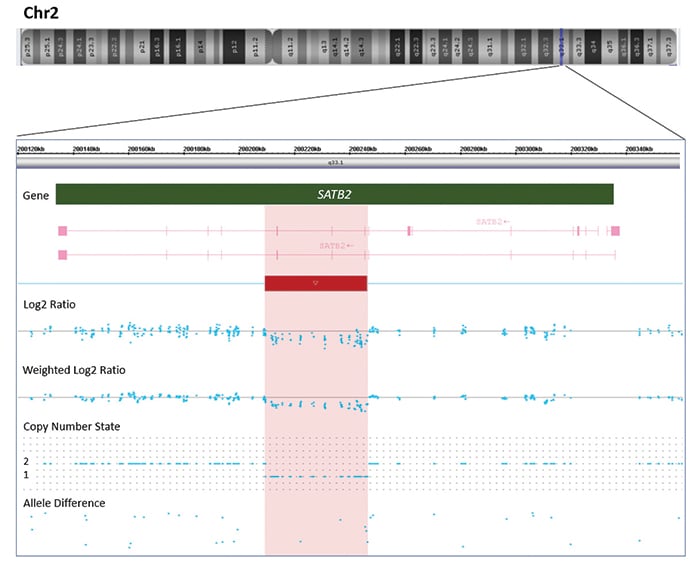
Genetic testing was arranged with informed consent. A buccal swab was collected from the patient, and DNA was extracted per standard protocols. Chromosomal microarray testing was performed to assess copy number variants using the CytoScan HD array (Thermo Fisher Scientific). It revealed an interstitial loss on the long arm of chromosome 2 at band q33.1, spanning approximately 35 kb in length and consisting of 88 markers/probes, arr[GRCh37] 2q33.1(200209972_200245277)x1 (Fig. 1B). This copy number loss spans 200209972_200245277 and includes exons 6–8 (NM_015265.4, hg19) of the Special AT-rich sequence-binding protein 1 (SATB2, OMIM #608148) gene (Fig. 2).
SATB2 is a DNA-binding protein involved in chromatin and transcriptional regulation. The deleted regions of the SATB2 protein include the CUT1 and CUTL domains, which are known hotspots for pathogenic single nucleotide missense variants in SATB2-associated syndrome (SAS). Given the strong overlap between the patient’s presentation with SAS and these molecular findings, the diagnosis of SAS was made based on the ACMG and Clinical Genome Resource joint consensus recommendations.1,2 While cascade genetic testing has not been performed, given that the patient has three healthy full siblings and both parents are unaffected, this variant is likely de novo. SAS is almost exclusively caused by de novo variants and has a germline mosaicism risk of 1.3 percent.3
Discussion. SATB2-associated syndrome, also known as Glass syndrome, is an autosomal-dominant neurogenetic multisystemic disorder caused by variants in Special AT-rich sequence-binding protein 1 (SATB2). It was first described in 1989 and mapped to the 2q32.2q33.1 region based on a report of an individual with severe developmental delay.4 The SATB2 gene encodes a nuclear matrix DNA-binding protein that participates in transcription regulation and chromatin remodeling by specifically binding to genomic nuclear matrix attachment regions. In 2003, the gene SATB2, which resides within the 2q33.1 region, was uncovered as causing cleft palate in two individuals with intellectual delay. Subsequent studies have confirmed that the dominant presenting symptom of SAS is intellectual disability/developmental delay (ID/DD). SAS is thought to represent 0.3 percent of individuals with ID/DD of unknown etiology.5,6 To elucidate a more explicit genotype-phenotype correlation by deconvolving the effects of SATB2-mediated effects versus those due other genes, a distinction has recently been drawn between individuals who harbor variants that affect only SATB2 versus larger variants that affect SATB2 and any surrounding genes. Of the patients with intragenic variants, the majority have single nucleotide variants, estimated to be approximately 68.3 percent. By contrast, intragenic deletions in SATB2 have been described much less frequently.3
SATB2-affecting variants are either large deletions encompassing SATB2 and surrounding genes, or smaller variants that involve only SATB2.3,7 Among the variants that involve only SATB2, the majority are single base substitutions and small indels (82.7 percent).3 Intragenic copy number variants of SATB2 constitute a minority of reported cases.3 Interestingly, in reviewing the literature, only 24 unique intragenic deletions have been reported to cause SAS3,8-13 (Fig. 2). In fact, some resources that compile variants in SATB2 do not include an option for copy number variants. Nevertheless, intragenic copy number variants are considered loss-of-function variants and have been associated with the same phenotypic presentation as single nucleotide variants.3
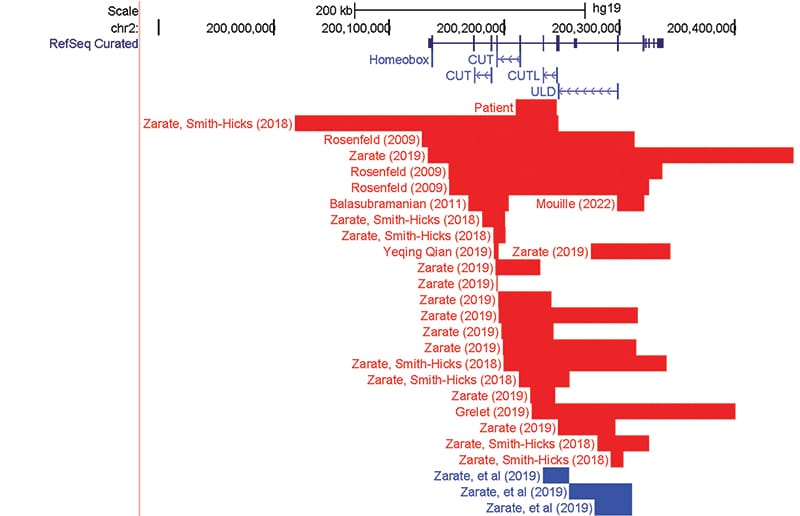
In conclusion, SAS is a rare neurogenetic disease with fewer than 300 reported cases that relies heavily on molecular testing for diagnosis. Most pathogenic intragenic variants are single nucleotide variants; however, increasingly small disease-causing deletions and duplications are being reported. We report an additional case of a two-year-old female with developmental delay and cleft palate who had a deletion spanning exons 6–8 detected on chromosomal microarray. In reviewing the literature, we noted that intragenic variants most frequently overlap with the CUT1, CUTL, and ULD protein domains, which is a similar distribution to the missense variants found in SAS. Intragenic deletions, particularly in exons 4 and 6–8, are a reoccurring disease-causing variant type in SATB2-associated syndrome, demonstrating the clinical utility of chromosomal microarray in detecting disease-causing variants in SAS.
- Miller DT, Adam MP, Aradhya S, et al. Consensus statement: chromosomal microarray is a first-tier clinical diagnostic test for individuals with developmental disabilities or congenital anomalies. Am J Hum Genet. 2010;86(5):749–764.
- Riggs ER, Andersen EF, Cherry AM, et al. Technical standards for the interpretation and reporting of constitutional copy-number variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics (ACMG) and the Clinical Genome Resource (ClinGen). Genet Med. 2020;22(2):245–257.
- Zarate, YA, Bosanko KA, Caffrey AR, et al. Mutation update for the SATB2 gene. Hum Mutat. 2019;40(8):1013–1029.
- Glass IA, Swindlehurst CA, Aitken DA, McCrea W, Boyd E. Interstitial deletion of the long arm of chromosome 2 with normal levels of isocitrate dehydrogenase. J Med Genet. 1989;26(2):127–130.
- Zarate YA, Smith-Hicks CL, Greene C, et al. Natural history and genotype-phenotype correlations in 72 individuals with SATB2-associated syndrome. Am J Med Genet A. 2018;176(4):925–935.
- Deciphering Developmental Disorders Study. Large-scale discovery of novel genetic causes of developmental disorders. Nature. 2015;519(7542):223–228.
- Zarate YA, Bosanko KA, Thomas MA, et al. Growth, development, and phenotypic spectrum of individuals with deletions of 2q33.1 involving SATB2. Clin Genet. 2021;99(4):547–557.
- Mouillé M, Rio M, Breton S, et al. SATB2-associated syndrome: characterization of skeletal features and of bone fragility in a prospective cohort of 19 patients. Orphanet J Rare Dis. 2022;17(1):100.
- Grelet M, Mortreux J, Alazard E, Sigaudy S, Philip N, Missirian C. SATB2-associated syndrome: first report of a gonadal and somatic mosaicism for an intragenic copy number variation. Clin Dysmorphol. 2019;28(4):205–210.
- Qian Y, Liu J, Yang Y, et al. Paternal low-level mosaicism-caused SATB2-associated syndrome. Front Genet. 2019;10:630.
- Asadollahi R, Oneda B, Joset P, et al. The clinical significance of small copy number variants in neurodevelopmental disorders. J Med Genet. 2014;51(10):677–688.
- Kaiser AS, Maas B, Wolff A, et al. Characterization of the first intragenic SATB2 duplication in a girl with intellectual disability, nearly absent speech and suspected hypodontia. Eur J Hum Genet. 2015;23(5):704–707.
- Liedén A, Kvarnung M, Nilssson D, Sahlin E, Lundberg ES. Intragenic duplication—a novel causative mechanism for SATB2-associated syndrome. Am J Med Genet A. 2014;164A(12):3083–3087.
- Aguilera C, Gabau E, Ramirez-Mallafré A, et al. New genes involved in Angelman syndrome-like: expanding the genetic spectrum. PLoS One. 2021;16(10):e0258766.
- Zarate YA, Vernon HJ, Bosanko KA, et al. Case report: SATB2-associated syndrome overlapping with clinical mitochondrial disease presentation: report of two cases. Front Genet. 2021;12:692087.
- Lo HY, Ng WF, Fong NC, Lui CYD, Lam CW. Novel finding of lissencephaly and severe osteopenia in a Chinese patient with SATB2-associated syndrome and a brief review of literature. Am J Med Genet A. 2022;188(7):2168–2172.
Dr. Hubler is a clinical pathology resident in the physician scientist training pathway, Department of Pathology and Immunology; Dr. Gooch is an assistant professor of pediatrics, genetics, and genomic medicine, Division of Genetics and Genomic Medicine, Department of Pediatrics; Erin Hediger is a clinical genetic counselor, Division of Genetics and Genomic Medicine, Department of Pediatrics; and Dr. Cao is an assistant professor of pathology and immunology, Division of Laboratory and Genomic Medicine, Department of Pathology and Immunology—all at Washington University School of Medicine in St. Louis.
Test yourself
Here are three questions taken from the case report.
1. What is the first-line diagnostic test for patients with intellectual disability and/or developmental delay?
a. Chromosomal microarray
b. Exon sequencing
c. M-Chat
2. Chromosomal microarray is designed to detect which type of variants?
a. Unbalanced chromosomal gains or losses
b. Low-level mosaicism
c. Single nucleotide variants
3. What is the most frequent variant described in SATB2-associated syndrome?
a. Missense variants
b. Large deletions
c. Internal tandem duplication
Answers are online now at www.amp.org/casereports.